Modified glucocorticoid stabilizes dysferlin-deficient muscle cell membrane in experimental models

Limb girdle muscular dystrophy type 2B (LGMD2B) – a disease so rare that researchers aren’t even sure how many people it affects – is characterized by chronic muscle inflammation and progressively weakened muscles in the pelvis and shoulder girdle. It can affect able-bodied people during their childbearing years and makes it difficult to tiptoe, walk, run or rise unaided from a squat. Ultimately, many with the muscle-wasting condition require wheelchair assistance. There is no therapy approved by the Food and Drug Administration for this condition.
In a head-to-head trial between the conventional glucocorticoid, prednisolone, and a modified glucocorticoid, vamorolone, in experimental models of LGMD2B, vamorolone improved dysferlin-deficient muscle cell membrane stability and repair. This correlated with increased muscle strength and decreased muscle degeneration, according to a Children’s-led study published online Aug. 27, 2018, in Molecular Therapy. By contrast, prednisolone worsened muscle weakness, impaired muscle repair and increased myofiber atrophy.
“These two steroids differ by only two chemical groups,” says Jyoti K. Jaiswal, MSC, Ph.D., a principal investigator at Children’s National Health System and senior study author. “One made muscle repair better. The other made muscle repair worse or about the same as untreated experimental models. This matches experience in the clinic as patients with LGMD2B experienced increased muscle weakness after being prescribed conventional glucocorticoids, such as prednisolone.”
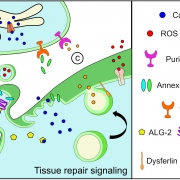
Healthy muscle cells rely on the protein dysferlin to properly repair the sarcolemmal membrane, a cell membrane specialized for muscle cells that serves a vital role in ensuring that muscle fibers are strong enough and have the necessary resources to contract. Mutations in the DYSF gene that produces this essential protein causes LGMD2B.
Jaiswal likens the plasma membrane to a balloon that sits atop the myofiber, a long cell that when healthy can flex and contract. If, in the process of myofiber contraction, the plasma membrane experiences anything out of sync or overly stressful, it develops a tear that needs to be quickly sealed. An intact balloon keeps air inside; tear it, and air escapes. When the plasma membrane tears, calcium from the outside leaks in, causing the muscle cell to collapse into a ball and die. The body contends with the dead cell by breaking it up into fragments and sending in inflammatory cells to clear the debris.

Lack of dysferlin is associated with increased lipid mobility in the limb girdle muscular dystrophy type 2B (LGMD2B) cell membrane, which is further increased by injury and prednisolone treatment, causing failure of these cells to undergo repair. By contrast, vamorolone treatment stabilizes the LGMD2B muscle cell membrane to near healthy cell level, enabling repair of injured cells.
The study team got the idea for the current research project during a previous study of the experimental treatment vamorolone for a different type of muscular dystrophy. “In Duchenne muscular dystrophy (DMD), treatment with vamorolone not only reduced inflammation, but the membranes of muscle fibers were stabilized. That was the team’s ah-hah moment,” he says.
Three different doses of vamorolone were tested on cells derived from patients with LGMD2B with higher cell membrane repair efficacy seen with rising treatment dose. The dysferlinopathic experimental models were treated for three months with daily doses of cherry syrup laced with either 30 mg/kg of vamorolone or prednisolone or cherry syrup alone as the placebo arm.
“Right now there are zero treatments,” he says. People with LGMD2B turn to rehabilitative therapies and movement aids to cope with loss of mobility. Doctors are cautioned not to prescribe steroids. Jaiswal says many patients with LGMD2B grew up doing strenuous exercise, former athletes whose first indication of a problem was muscle cramping and pain. How this progresses to muscle weakness and loss is an area of active research in Jaiswal’s lab. “While additional research is needed, our findings here suggest that modified steroids such as vamorlone may be an option for some patients,” Jaiswal says.
“There is a nuance here: In addition to genomic effects, steroids also have physical effects on the cell membrane which may make some of the approved steroids ‘good’ steroids for dysferlinopathy that could selectively be used for this disease,” adds Sen Chandra Sreetama, lead study author. Further research could indicate whether vamorolone, which is in Phase II human clinical trials for DMD, or any off-the-shelf drug could slow decline in muscle function for patients with LGMD2B.
Additional Children’s study authors include Goutam Chandra; Jack H. Van der Meulen; Mohammad Mahad Ahmad; Peter Suzuki; Shivaprasad Bhuvanendran; and Kanneboyina Nagaraju and Eric P. Hoffman, both of ReveraGen BioPharma.
Research reported in this news release was supported by the Clark Charitable Foundation; Muscular Dystrophy Association, under award number MDA277389; National Institute of Arthritis and Musculoskeletal and Skin Diseases, under award number R01AR055686; National Institutes of Health (NIH), under award numbers K26OD011171 and R24HD050846; and the District of Columbia Intellectual and Developmental Disabilities Research Center under NIH award number 1U54HD090257.