This prestigious award is presented annually to an individual in recognition of a lifetime of achievement in pediatric nephrology. Dr. Moxey-Mims joins a distinguished group of recipients for their clinical excellence, teaching and advocacy.
Her decades of service to colleagues, mentees and the thousands of children whose lives she has touched are celebrated through this recognition, both at the national level and within the Children’s National Hospital community.
“I am deeply honored to receive the Henry L. Barnett Award,” Dr. Moxey-Mims said. “It is a reminder of the privilege and responsibility of caring for children, advancing pediatric nephrology and guiding the next generation of clinicians through mentorship and teaching.”
About Dr. Marva Moxey-Mims
Dr. Moxey-Mims has served as the chief of Nephrology since 2017, providing visionary leadership and unwavering support to faculty and staff. Known for her inclusive and collaborative approach, she is recognized for her commitment to mentoring new faculty and fellows, helping them navigate their roles by being a source of information and direction.
Before stepping into her current role at Children’s National, Dr. Moxey-Mims served as deputy director for clinical science in the Division of Kidney, Urologic and Hematologic Diseases at the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) at the National Institutes of Health. She earned her undergraduate degree from McGill University and medical degree from Howard University, followed by her pediatric residency and clinical nephrology training at Children’s National.
With more than 125 scientific publications, Dr. Moxey-Mims has made substantial contributions to the understanding of glomerular disease and chronic kidney disease (CKD) in children. The study, which she initiated while at the NIDDK, remains the largest study of pediatric CKD ever conducted in North America.
As chief, Dr. Moxey-Mims has built a dedicated, multidisciplinary team, while fostering a culture that encourages both clinical excellence and research innovation. She oversees a robust kidney transplant program, pediatric dialysis center and a growing number of specialized care programs, all aimed at improving outcomes for children with kidney disease. Under her leadership, the Children’s National Division of Nephrology was recognized again this year as one of the top pediatric nephrology programs in the nation by U.S. News and World Report.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2026/04/MMM-Award-feature.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2025/09/InnovationDistrict_CN_WebHeader-1396px-1030x151.pngInnovation District2026-04-27 10:46:492026-04-27 10:48:27Marva Moxey-Mims, MD, receives Henry L. Barnett Award
Andrew Dauber, MD, MMSc, chief of Endocrinology, and Denver Brown, MD, pediatric nephrologist, were featured as guests on The Kidney Chronicles: A Pediatric Nephrology Podcast. The episode focused on growth hormone in chronic kidney disease (CKD).
“As a pediatric nephrologist, it’s hard not to be interested in growth since growth failure and short stature is such a common finding in pediatric CKD, so I like to think all of us providers are interested in growth,” Dr. Brown said.
Both experts provided insight on common challenges of CKD patients, like when to start growth hormone treatment, how the condition can progress through metabolic acidosis and even the social impacts of common symptoms on everyday life. The episode gave a holistic view of how endocrinology and nephrology work together to support patients with CKD.
“While growth hormone is an important option, and I think an important treatment modality for kids who aren’t growing well with CKD, it’s just as important for families to think about other coping mechanisms for their kids,” Dr. Dauber said. “And that’s true for anyone with chronic disease.”
Listen to the full episode, “Growth hormone in CKD,” on PodBean or Spotify.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2026/03/The-kidney-chronicles-feature.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2025/09/InnovationDistrict_CN_WebHeader-1396px-1030x151.pngInnovation District2026-03-30 16:15:102026-03-30 16:16:21In the news: Growth hormone in CKD
The final paper includes clear, unified guidance on how to recognize high blood pressure in children and adolescents early and manage this condition throughout lifetimes to improve long-term cardiovascular and kidney disease outcomes.
Hypertension in children has been under-recognized and under-researched, particularly when compared to hypertension in adults. The lack of global consensus on diagnostic thresholds and recommended evaluations for children, along with the resource limitations in many areas of the world leading to limited clinical and community-based data, all contribute to existing knowledge gaps.
In a recent position paper from the International Society of Hypertension (ISH), a global team of experts, including new Children’s National Hospital hypertension expert, Tammy Brady, MD, PhD, combined years of research and expertise to provide important recommendations to clinicians caring for children with hypertension in diverse healthcare settings.
The final paper includes clear, unified guidance on how to recognize high blood pressure in children and adolescents early and manage this condition throughout lifetimes to improve long-term cardiovascular and kidney disease outcomes.
The big picture
The overarching goal of the paper is to facilitate early diagnosis and more precise management. Throughout, many areas of pediatric hypertension are addressed, from epidemiology and diagnostic evaluation to blood pressure measurement techniques and lifestyle and pharmacologic treatment.
While it reflects the most current research and evidence-based guidance, the paper also provides actionable resources that can be implemented across a wide range of clinical settings, including web-based educational links, clear graphics and step-by-step guides to support accurate measurement, diagnostic evaluation and treatment decisions.
One of the most compelling aspects of this work is the incorporation of longitudinal evidence showing that elevated blood pressure in childhood directly increases the risk of major cardiovascular and kidney events in adulthood, Dr. Brady said.
“Rather than merely revising previous recommendations, the paper reframes pediatric hypertension as a life-course condition that demands early, coordinated intervention to meaningfully change cardiovascular risk trajectories,” Dr. Brady added.
Moving the field forward
This paper advances the field in several important ways, most notably its broad relevance, focusing on regional differences in resources and practice. Not only does it focus on realistic, everyday practices for clinicians, but the paper also draws expertise from 12 countries, making it globally applicable.
It brings together prevention, diagnosis, treatment and health system considerations into a cohesive framework, extending beyond individual patient care to address policy and public health implications.
What’s next?
Dr. Brady sees the foundation for this paper expanding through her research and clinical efforts at Children’s National, positioning the hospital as a global leader in pediatric hypertension and cardiovascular risk research.
On the research front, Dr. Brady’s work will focus on refining pediatric blood pressure measurement standards, deepening understanding of early cardiac and vascular remodeling and advancing device validation, particularly for special populations such as children and individuals with obesity.
Clinically, she emphasizes the importance of strengthening implementation science to ensure that evidence-based screening and management strategies are adopted consistently and sustainably across care settings.
“Publication is only the first step,” Dr. Brady said. “The real impact will come from adoption and sustained application.”
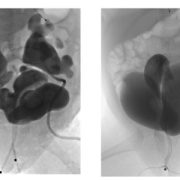
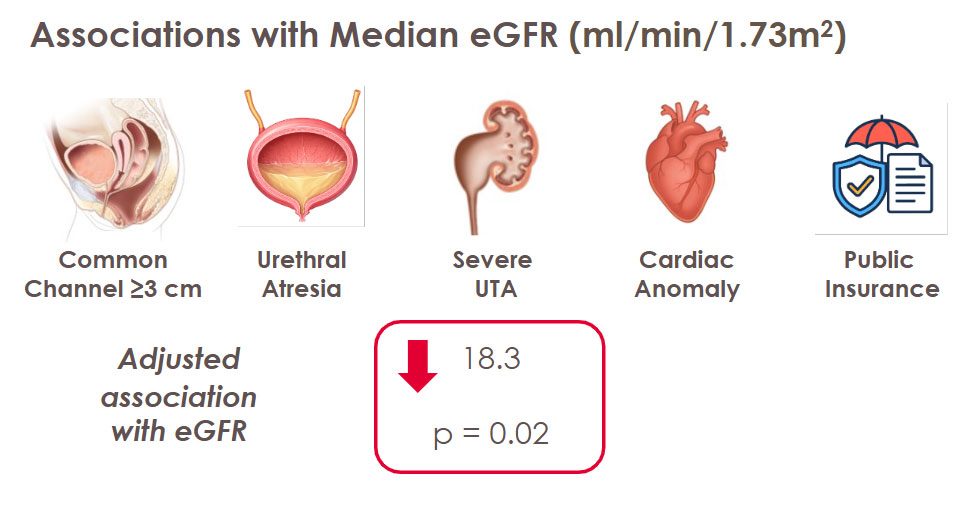
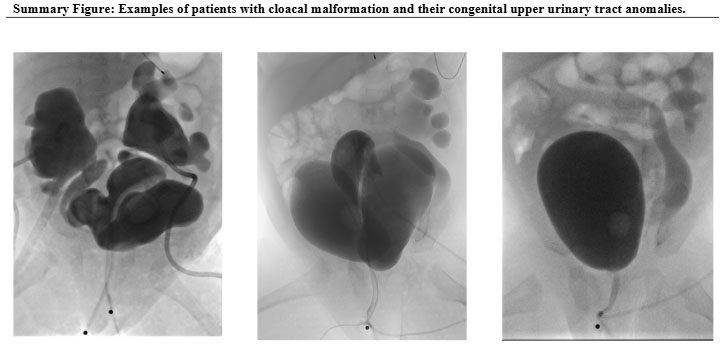
Children born with cloacal malformations face many medical challenges beyond surgery. One major concern is kidney health, which may not always be obvious at first. In a recent study published in the Journal of Pediatric Urology, experts at Children’s National Hospital found that many of these children show early signs of kidney problems, often related to abnormalities in the kidneys or urinary tract that are present from birth.
How does this work move the field forward?
In the past, doctors did not fully understand why some children with cloaca develop chronic kidney disease (CKD). Earlier studies showed that about 6% of children with complex cloaca needed a kidney transplant by around age 10, but the exact cause of their kidney problems was unclear.
“Our study shows that some kidney damage in children with cloaca actually begins before birth and is linked to problems in the kidneys or urinary tract that are present from the start,” said Briony Varda, MD, MPH, urologist in the Division of Urology and the Division of Colorectal and Pelvic Reconstruction at Children’s National. “This early damage may reduce a child’s kidney reserve, but it may not show up on common blood tests like serum creatinine.”
By using newer, more accurate ways to measure kidney function, such as the CKID U25 GFR formula, researchers were able to detect hidden kidney problems that standard tests might miss. This helps doctors identify at-risk children earlier and take steps to protect their kidney health over time.
What did you find that excites you?
The study found that children with cloacal anomalies can have kidney problems from birth, even when routine tests look normal.
“These findings raise important questions for care,” Dr. Varda said. “Can careful management slow kidney damage, or is it set before birth? How will these early kidney issues affect patients as they grow?”
Understanding these answers could help doctors plan better long-term care and protect kidney health throughout childhood and beyond.
How is Children’s National leading in this space?
Children’s National has performed more primary cloaca repairs than any other hospital in the U.S. over the past five years, according to national data.
“Our team works closely in the operating room and with many specialists outside the OR,” Dr. Varda said. “For this study, our kidney expert, Melissa Meyers, MD, contributed her guidance. Since 2020, we’ve also maintained a careful database that tracks outcomes for this rare condition. This work helps us learn and improve care for every patient.”
https://innovationdistrict.childrensnational.org/wp-content/uploads/2026/02/cloacal-malformation-feature.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2025/09/InnovationDistrict_CN_WebHeader-1396px-1030x151.pngInnovation District2026-02-25 11:34:202026-02-25 11:36:26New study reveals hidden kidney dysfunction in children with cloacal malformations
Two simple ultrasound measurements can help doctors better spot kidney blockages in children and reduce the need for radiation tests.
Kidney blockages in children, often caused by ureteropelvic junction obstruction (UPJO), can be hard to diagnose. These blockages can lead to hydronephrosis, or swelling of the kidney, which can damage kidney function if not found early.
The most accurate test, called a MAG3 scan, shows how well the kidney works and drains urine. However, this test uses radiation and requires an IV. For that reason, doctors often use ultrasound first since it is safe and does not use radiation. The challenge is that reading ultrasounds to judge how serious the swelling is can be unclear and based on opinion.
Doctors use grading systems to describe the severity of hydronephrosis, or kidney swelling. These systems often depend on whether the kidney tissue appears “thin.” The problem is that there is no clear rule for what “thin” actually means. As a result, different doctors may describe the same ultrasound differently, which can make it harder to decide which children need more testing.
How does this work move the field forward?
In this study, Dr. Krill and his team found that two ultrasound measurements —the width of the kidney’s central area (renal pelvic diameter) and the thickness of the kidney tissue (renal parenchymal thickness) — were linked to signs of blockage seen on a MAG3 scan.
Researchers looked at whether different radiologists would get the same results when making these measurements. They found very strong agreement between doctors, even when they used slightly different ways to find the thinnest part of the kidney tissue.
“We’ve relied on visual grading for years,” Dr. Krill said. “We wanted to see if we could use clear measurements that are repeatable and directly connected to how the kidney is functioning.”
The results showed that these measurements are reliable and can be standardized.
How will this work benefit patients?
Using these clear measurements can help doctors better identify which children with hydronephrosis are truly at risk for blockage. This means fewer children may need a MAG3 scan if their ultrasound measurements show they are low risk. At the same time, children who are at higher risk for blockage can be identified sooner. This approach supports safer, more personalized care.
How is Children’s National leading in this space?
Children’s National is one of the first groups to connect clear ultrasound measurements with objective results from MAG3 scans. By combining these pieces, the team is helping move care away from opinion-based grading and toward more consistent, measurement-based decision-making. This work supports smarter testing and more tailored follow-up for each child.
After 21 years at Johns Hopkins Medicine, where she most recently served as the Vice Chair for Clinical Research in the Department of Pediatrics and Medical Director of the Pediatric Hypertension Program, Children’s National is excited to welcome Dr. Brady and her vast knowledge in the fields of pediatric hypertension and clinical research.
Dr. Brady’s previous research highlights her commitment to creating positive health outcomes for children. With expertise in hypertension, particularly obesity-related hypertension, blood pressure (BP) measurement, BP device accuracy and healthy lifestyle education, Dr. Brady will bring a unique and innovative set of expertise to the team.
“Our Nephrology division is very excited about the world-renowned clinical hypertension expertise that Dr. Brady will be adding to our team,” Marva Moxey-Mims, MD, chief of the Division of Nephrology, said.
At Children’s National, Dr. Brady joins the Center for Health Outcomes and Delivery Science and will lead the Grants Enhancement Program as well as a newly created Hypertension and Kidney Health Research Laboratory. Additionally, she plans to launch a new hypertension clinic, where she hopes to further study risk factors for cardiovascular disease in children.
“We are thrilled to welcome Dr. Brady to Children’s National and the Center for Health Outcomes and Delivery Science community,” Beth A. Tarini, MD and Monika Goyal, MD, co-directors of the center, said. “She brings deep expertise and a strong commitment to mentorship and collaboration. Her leadership will strengthen our ability to support investigators and advance research that improves the lives of children and families.”
In a recent Q&A, Dr. Brady highlighted her research and professional background, along with her excitement stepping into her new role.
Q: How did your previous work prepare you for this role?
A: I have been so fortunate to train with and be mentored by the most amazing physician scientists who are not only experts in their field but are passionate about their work. Starting from residency when Dr. Joseph Flynn mentored me on my very first research project, investigating clinical and demographic predictors of increased cardiovascular disease risk in children, to working with Dr. Larry Appel and others on a suite of randomized clinical trials testing the importance of various steps on BP measurement accuracy, I have learned so much. I have personally conducted all aspects of clinical research, from manual chart review and data abstraction (with paper charts!) to recruitment and retention of pediatric participants in cross-sectional and longitudinal studies. These experiences have enabled me to be an effective research staff supervisor (I never ask staff to do anything I haven’t already done!) and mentor.
In my former roles at Johns Hopkins, I focused on making research easier by helping trainees and faculty navigate every aspect of research, including study design, grant writing, budget development, funding acquisition, study implementation and results dissemination. I look forward to bringing this expertise to my new roles within the Center for Health Outcomes and Delivery Science at Children’s National.
While I have also had significant didactic training in pediatric nephrology, epidemiology and clinical investigation, my “boots on the ground” experience in research and clinical care has had the greatest impact on my personal and professional development. Medicine and research are team sciences, and I strongly believe we are only able to provide the best care when we collaborate with others who bring different but complementary expertise and life experiences.
Q: You’ll be helping to launch and lead a new hypertension program at Children’s National. Can you tell us more about that vision? What is unique about this type of program?
A: The hypertension program at Children’s National will be very family-focused, individualized and multidisciplinary. In addition to completing a full evaluation that includes “old school” BP measurements with a manual cuff and stethoscope according to clinical practice guidelines, each patient will undergo a thorough and personalized assessment to provide the correct diagnosis and evaluate for causes of hypertension and other conditions that could increase their cardiovascular disease risk.
All patients and their families will be counseled on learned heart healthy behaviors that preserve cardiovascular health. Habits like eating well, moving daily, getting good sleep, minimizing screen time, avoiding nicotine and minimizing stress are good for ALL people, with and without high BP. I often tell families: it’s easier to learn a new behavior than unlearn an unhealthy behavior, so the earlier we promote healthy behaviors the better!
The hypertension program will incorporate established and emerging assessments to provide a robust picture of each child’s cardiovascular risk. It will include out-of-office BP assessments through 24-hour ambulatory BP monitoring and home BP monitoring. It is key to know what a person’s BP is like out of the office to eliminate any contribution of the “white coat effect” prior to embarking on an extensive evaluation and treatment plan. It will also incorporate heart imaging and measures of vascular stiffness and reactivity to paint a more holistic picture of their cardiovascular health. I will also be working closely with the Improving Diet, Energy and Activity for Life (IDEAL) Clinic seeing youth who are followed there and are found to have high BP. By seeing kids in concert with IDEAL experts, I hope to make things easier for families.
Q: Share a little about your research portfolio. What is your particular research passion?
A: I am very passionate about cardiovascular health promotion, and as such, that is where my research has focused. Early on, I investigated how to best determine which children with hypertension were more likely to have an early form of cardiovascular disease called left ventricular hypertrophy (LVH). This work revealed that obesity was a significant risk factor for LVH, and in fact, when I followed hypertensive children and adolescents over a 12-month period of time I found that those who gained weight were the ones who had their heart remodel in an unhealthy way, despite having good BP control.
I was awarded a National Institutes of Health grant to test the effect of a behavioral intervention using Instagram on weight and BP among children with obesity. Over six months, kids following the Instagram account gained less weight than those who were in the control group – suggesting that increased touch points and check ins can improve cardiovascular health. This led me to develop an obesity hypertension clinic to not only take better care of children with obesity and hypertension in the here and now – with more frequent follow up and multidisciplinary care – but also to serve as a means to learn how to take better care of children in the future.
All the children in this clinic were invited to join a clinical research registry which led to more discoveries: sleep apnea, anxiety and depression are highly prevalent in kids with obesity and hypertension – and when present, amplify the risk for high BP and LVH. I also discovered that many triage BPs – the ones taken prior to seeing the doctor – were inaccurate in youth with obesity. Because so many decisions are often made based on these measurements, optimizing triage BP measurement is key to improving clinic efficiency and minimizing unnecessary follow up and evaluation in children who have a high BP related to technique.
My latest efforts have focused on exploring the importance of established BP measurement steps on measurement accuracy. I led four randomized trials in adults to determine how important it is to rest for five minutes prior to BP measurement, to have BP measured in a quiet, private setting, to select a cuff size based on measured arm size, and to rest the arm on a table next to you positioned with the cuff at heart level. We tested these conditions because these are steps that are often not followed and add a significant amount of time to the measurement procedure, impacting clinic flow. We found that for adults without high BP, there is (a) no need to wait prior to measurement, (b) a loud, non-private measurement setting doesn’t significantly impact BP accuracy, (c) it is essential to select the correct cuff size, as choosing too small a cuff can overestimate BP by up to 20mmHg, and (d) having the arm rest on a desk makes a difference, as not adhering to this can overestimate BP by almost 10 mmHg. I am now hoping to test these conditions in kids – the easier we make BP measurement, the more kids we will be able to effectively screen for high BP!
Q: What stood out to you when deciding to join Children’s National? And what are you most excited to bring to your new role?
A: What made me most excited to join Children’s National are the people. Not only are there phenomenal physician scientists here, but every single member of the Children’s family has been so kind, welcoming and committed to providing excellent care to the children and families we serve.
The opportunity to work with Drs. Goyal and Tarini is a dream. They not only conduct meaningful, practical, creative and impactful research, but they are visionaries. I am excited to collaborate with them in the center and learn from them as I grow the Grants Enhancement Program. Similarly, I am so excited to work with Dr. Moxey-Mims. She is a tireless advocate for children with kidney disease and has expansive expertise in how to conduct meaningful clinical research in this population.
Q: What keeps you passionate about the work you do?
A: The families who trust me to care for their children. The relationships I have formed with my patients along with the opportunity to celebrate their successes and guide them through their challenges is something I cherish. Learning what is important to the families I care for allows me to focus my work in a way that it is impactful for them.
I also just really love clinical research. One of my favorite things to do is meet with trainees and junior faculty, learn about what their clinical and research interests are, and then start drawing conceptual models and directed acyclic graphs. I won’t even get into the thrill I get from writing Stata code – it’s all really just so much fun.
Q: Last question – what do you like to do with your time outside of work?
A: I love travel, live music, Boston College football games and FaceTiming my kids.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2026/02/Tammy-Brady-Feature.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2025/09/InnovationDistrict_CN_WebHeader-1396px-1030x151.pngInnovation District2026-02-13 13:57:412026-02-17 10:22:24Welcoming pediatric hypertension expert Tammy M. Brady, MD, PhD to Children’s National
Kidney failure is relatively rare in children compared to adults, making it more difficult for individual dialysis centers to conduct large-scale studies.
The ability to recognize nutritional gaps in children with kidney disease is essential to effective care planning and long-term condition management. Pediatric patients on dialysis are a particularly fragile population when it comes to nutritional risks, often facing poor appetite, gastrointestinal symptoms and dietary restrictions that make adequate intake challenging to maintain.
In a recent study, experts at Children’s National Hospital addressed a gap in pediatric kidney care by examining whether normalized protein catabolic rate (NPCR), which measures daily dietary protein intake, could be used as a reliable tool to identify compromised nutritional status in pediatric hemodialysis (HD) patients ages 0-12 years old. While NPCR had been studied in adolescents, its usefulness in younger children had not previously been established.
This study will serve as the foundation for future research, with plans already underway to expand this work across multiple centers.
The big picture
The analysis consisted of 758 observations of 58 patients and evaluated nutritional risk using a composite indicator of compromised nutritional status, including low body weight, poor growth, low appetite and biochemical abnormalities. Results of this single-cohort study showed that pediatric HD patients with an NPCR level less than 1.2 had two-fold increased odds of poor nutritional status.
When broken down by age, children 4-12 years-old showed an even stronger association, with four-fold increased odds of compromised nutritional status. This relationship was less clear among infants and toddlers ages 0-3, highlighting an important area for continued research.
The patient impact
Tools to assess nutritional status in young children on dialysis are very limited. As the first study to establish NPCR as a marker of nutritional status for chronic HD patients ages 0-12 – rather than solely focusing on adolescents – this research provides clinicians with a clearer, evidence-based NPCR threshold to help guide interventions for this population.
This guidance is critical during key periods of growth and development and has the potential to improve outcomes as children prepare for kidney transplantation.
Moving the field forward
Kidney failure is relatively rare in children compared to adults, making it more difficult for individual dialysis centers to conduct large-scale studies. As a result, pediatric research in this space is often limited by sample size. This study provides the methodological and clinical foundation for future work.
Building on these findings, leaders at Children’s National are collaborating through the Pediatric Nephrology Research Consortium to coordinate the first multi-center study of NPCR in pediatric HD patients. A larger sample size will allow for more definitive conclusions, particularly for the youngest patient cohort.
Kidney disease is a leading cause of mortality in SCD, underscoring the urgent need for earlier and more precise diagnostics.
“We’ve watched as silent kidney damage progresses year after year, often going undetected until significant harm has occurred,” said Andrew Campbell, MD, director of the Comprehensive Sickle Cell Disease Program at Children’s National and co-investigator of the study. “By identifying kidney disease earlier, we can intervene sooner with more effective disease-modifying therapies and curative treatments, potentially preventing or slowing progression to kidney failure.”
From discovery to impact
“We measure damaged mitochondria shed by the kidney in a few milliliters of urine so we can develop an early warning system for the onset of kidney damage,” said Marissa Howard, PhD, systems biologist and research fellow at George Mason University and principal investigator of the study. “Our partners at Children’s National are known for identifying innovative approaches to treat SCD. This exciting collaboration is collectively motivated by our ultimate goal of reducing the high mortality rate caused by Sickle Cell kidney failure in children affected by this disease.”
By translating cutting-edge science into clinical impact, Children’s National continues to advance next-generation diagnostics that improve outcomes for children with SCD and may extend to other diseases linked to mitochondrial dysfunction.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2023/02/diseased-kidneys-feature.png300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2025/09/InnovationDistrict_CN_WebHeader-1396px-1030x151.pngInnovation District2026-02-04 12:18:562026-02-04 12:21:53Advancing early detection for kidney disease in sickle cell
Children’s National is ranked one of the top 10 pediatric hospitals in the nation by U.S. News & World Report. Our faculty and staff are proud of the impact made on the lives of children and families in our community. Your participation in the U.S. News & World Report annual reputational survey validates the quality of care we provide and reflects the mutual respect and trust we share as healthcare professionals.
How to determine your voting eligibility
Voting for the U.S. News & World Report Best Children’s Hospitals rankings can be done only through Doximity.
To participate, physicians must:
Be board-certified and meet the eligibility criteria for the voting categories.
For child and adolescent psychologists, your account must be up to date with your specialty and subspecialty correctly marked.
Be a credential-verified member of Doximity (you must have an active and claimed Doximity profile).
Have all certifications and board documents currently up-to-date in your Doximity profile.
You have to claim your profile on Doximity.com to participate in the online survey. If you have not yet claimed your Doximity profile, go to Doximity.com, and click “Find My Profile.”
Once your profile has been claimed, you must confirm your email address and board certifications.
Verified Doximity members will receive an email inviting them to participate in the U.S. News survey.
For more information on how to claim your profile, visit Doximity.com
How to update and verify existing Doximity account information
Your Doximity profile must have up-to-date licenses, certifications and board documents.
Once you are logged in, your profile will automatically be in “Edit Mode.” You are able to add new items or edit existing information.
Update your Doximity profile and ensure your information is current.
Once registered, users wishing to participate in the online survey should:
Watch for an email from Doximity about the annual member survey.
Even if you don’t see the email, if you are a registered Doximity user, you can still vote by logging in to Doximity.com with your username and password during the voting period.
Once logged in, look for a U.S. News graphic or button on the homepage and click on it.
The survey asks users to name the hospitals that provide the best care in your respective specialty, without consideration to location or cost. Pediatric specialists will list 10 hospitals. The order in which you list the hospitals does not matter.
Please note: Children’s National Hospital is listed as “Children’s National Hospital Washington, DC” on the survey.
Visit Doximity’s FAQs if you have issues or questions about registration or claiming your profile.
How to cast your vote
In February 2026 when voting opens, all survey-eligible physicians will receive a notification on the Doximity app for Android or iOS. If you do not use the Doximity app, you will receive an email when voting opens.
Log in to your Doximity account at doximity.com or via the mobile app.
Click the Notifications icon or tap the “Submit your Nominations” button on the homepage. You can also search for “U.S. News Best Hospitals”
Select 10 hospitals in your respective specialty that you believe provide the best care in the United States.
Submit your vote
Having technical issues?
If you have difficulty registering with Doximity or completing the survey, please visit Doximity support for assistance.
Vote
The 2026 U.S. News & World Report Best Children’s Hospitals reputation voting will open in mid-February. Look for your Doximity notification to vote.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2026/01/SB106139-featured.png7501000Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2025/09/InnovationDistrict_CN_WebHeader-1396px-1030x151.pngInnovation District2026-01-12 11:24:572026-01-12 11:24:57Leading the way in pediatric kidney genetics innovation
In 2025, Innovation District readers gravitated toward stories that explored how research and clinical innovation are reshaping pediatric care in real time. This year’s most popular articles highlighted advances in complex surgical care, evidence-based treatments for chronic and neuropsychiatric conditions and emerging technologies — from wearable data to artificial intelligence — that are changing how clinicians diagnose, treat and support children and families. Read on for our list of the most popular articles we published on Innovation District in 2025.
The Cervical Spine program at Children’s National Hospital is responsible for treating a range of conditions, including trauma, congenital abnormalities and tumors. These conditions can lead to instability or misalignment of the cervical spine. “There are unique challenges in pediatric cases due to anatomical differences. The cervical spines of children are more at risk for injury because of their developmental stage and structural characteristics,” says Matthew Oetgen, MD, MBA, chief of Orthopaedic Surgery and Sports Medicine at Children’s National. (2 min. read)
Increasing evidence-based treatment is a key component of the Addictions Program at Children’s National Hospital, created in 2022 and led by Sivabalaji Kaliamurthy, MD. “We really want to focus on intervening in an evidence-based manner in the primary care setting because that is where most of our patients are going to first access care outside of the emergency room,” explains Dr. Kaliamurthy. (3 min. read)
For many children with short stature and other rare genetic growth disorders, there have been no next steps after usual treatment options prove ineffective. Researchers at Children’s National Hospital are digging deeper to find the root genetic causes of short stature disorders and creating novel, nuanced treatment options that have the opportunity to change how the field approaches these cases. (4 min. read)
Denver D. Brown, MD, nephrologist at Children’s National, is looking at whether untreated metabolic acidosis could potentially contribute to cardiovascular outcomes in children with chronic kidney disease (CKD). Here, she explains her motivation, findings and future directions for this critical research. (3 min. read)
A multidisciplinary therapy model developed at Children’s National shows promise for children with PANS and PANDAS, significantly reducing symptoms through structured cognitive-behavioral therapy and family-centered care. The approach could offer a new standard for treating these rare, complex neuropsychiatric disorders. (2 min. read)
A study from Children’s National reveals that common wearable devices like Fitbits may hold the key to improving how we identify Attention-Deficit/Hyperactivity Disorder (ADHD) in adolescents. By analyzing patterns in heart rate, activity levels and energy expenditure, researchers were able to predict ADHD diagnoses with striking accuracy, offering a glimpse into a future where objective, real-time data supports earlier and more personalized mental healthcare. (2 min. read)
A novel implantable pacemaker designed specifically for infants has demonstrated safety and effectiveness in stabilizing heart rhythms for at least two years. The multi-center study of 29 infants showed stable pacing, normal electrical parameters and expected battery life, offering a viable alternative to standard-size devices for the smallest children. (2 min. read)
Children who had heart surgery and come from less advantaged neighborhoods in the Washington, D.C., region are much more likely to die in the long term than those from neighborhoods with more wealth and opportunity. The finding was part of a presentation titled, Socioeconomic Disadvantage Is Associated with Higher Long-Term Mortality After Cardiac Surgery, by Jennifer Klein, MD, MPH, cardiologist at Children’s National Hospital, during the Society of Thoracic Surgeons Annual Meeting in Los Angeles. (2 min. read)
Experts from Children’s National traveled to Uganda to continue work on a pilot program applying artificial intelligence (AI) to the diagnosis of rheumatic heart disease (RHD). The team created a tool that uses AI to predict RHD by identifying leaky heart valves on handheld ultrasound devices, then prompts a referral for a full echocardiogram. (2 min. read)
Food insecurity is rising in Washington, D.C. and it’s hitting families with children the hardest. That’s why Children’s National Hospital created the Family Lifestyle Program (FLiP) – a multi-layered intervention, which offers Patient Navigation (FLiP-PN) and a Produce Prescription Intervention (FLiPRx). FLiP is a Food Is Medicine, clinical-community initiative that helps families get access to fresh food, build healthy habits and lower their risk of diet-related diseases like diabetes and obesity. (3 min. read)
https://innovationdistrict.childrensnational.org/wp-content/uploads/2025/12/2025-with-lightbulb-CNRI.jpg385685Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2025/09/InnovationDistrict_CN_WebHeader-1396px-1030x151.pngInnovation District2025-12-24 15:07:522025-12-24 16:43:36The best of 2025 from Innovation District
In 2025, Children’s National Hospital was featured in major national news outlets for pioneering advances in pediatric care, groundbreaking clinical research and powerful human stories of healing and hope. From gene therapy for sickle cell disease and innovative pacemakers for newborns to breakthrough transplants, cancer trials and emerging mental health concerns like AI psychosis, these stories highlight the hospital’s leadership across the full spectrum of pediatric medicine. The following ten highlights showcase the patients, families and experts behind this impact, as reported by outlets including NBC News, The Washington Post, Good Morning America, USA Today, Healio, ABC News and ESPN.
Children’s National patient Wedam, 19, begins the first steps for intensive gene therapy for sickle cell disease, discussing his skepticism while his mother expresses her joy and gratitude for the treatment. (NBC News)
Charles Berul, MD, pediatric electrophysiologist and emeritus chief of Cardiology, discusses his study highlighting the safety and efficacy of an innovative smaller pacemaker designed for newborns with critical congenital heart disease. (Healio)
Catherine Bollard, MBChB, MD, senior vice president and chief research officer, and the NexTGen team are poised to recruit patients for a new clinical trial that will take on an old, implacable foe: children’s solid tumors. (The Washington Post)
Yves d’Udekem, MD, PhD, chief of Cardiac Surgery, talked to Good Morning America about how an 11-year-old’s groundbreaking partial heart transplant will change his life and the lives of other children in need of valve replacements. (Good Morning America)
The Lilabean Foundation along with Brian Rood, MD, medical director of the Brain Tumor Institute, talked about how patients like Kasey Zachman are the motivation behind finding a cure for brain cancer. (ABC News)
USA Today Sports spoke with Gavin Brown and his parents, as well as Yi Shi, MD, a pediatric nephrologist at Children’s National Hospital, about their kidney transplant journey. (USA Today)
After Jayden Daniels visited Commanders fan Sarah Addison at Children’s National Hospital while she was being treated for myeloid leukemia, they quickly became friends. (ESPN)
A baby boy in Maryland is back home after being given a second chance at life, just before his first birthday. The boy’s mother and his surgeon, Manan Desai, MD, share the remarkable story of a moment that changed all of their lives. (NBC4)
Ashley Maxie-Moreman, PhD, clinical psychologist, spoke to ABC7 about what AI psychosis is and what parents need to know. (ABC7)
https://innovationdistrict.childrensnational.org/wp-content/uploads/2025/12/Logo-Collage-2025-feature.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2025/09/InnovationDistrict_CN_WebHeader-1396px-1030x151.pngInnovation District2025-12-23 13:53:592025-12-31 09:09:17Children’s National in the News: 2025
Children’s National Hospital in Washington, D.C., was ranked as a top hospital in the nation by the U.S. News & World Report 2025-26 Best Children’s Hospitals annual rankings. This marks the ninth straight year Children’s National has made the Honor Roll list. The Honor Roll is a distinction awarded to only 10 children’s hospitals nationwide.
For the fifteenth straight year, Children’s National ranked in 10 specialty services and is the highest U.S. News ranked children’s hospital in Washington, D.C., Maryland and Virginia. Last year, U.S. News introduced pediatric & adolescent behavioral health as a service line in its rankings. While there are no ordinal rankings for behavioral health, the Children’s National program was named one of the top 50 programs in the country for the second year in a row.
“To be named among the nation’s top children’s hospitals for nine years in a row is a reflection of the extraordinary expertise, innovation and heart that our teams bring to every child and family we serve,” said Michelle Riley-Brown, MHA, FACHE, president and chief executive officer of Children’s National. “Our leadership in specialties like neurology, cancer, and diabetes and endocrinology underscores the national impact of our work, and we remain focused on setting new standards in pediatric care.”
The annual rankings are the most comprehensive source of quality-related information on U.S. pediatric hospitals and recognizes the nation’s top 50 pediatric hospitals based on a scoring system developed by U.S. News.
“Being a top-ranked pediatric hospital means more than just excelling in a single specialty — it means being a pillar of outstanding care for your entire region,” said Ben Harder, chief of health analysis and managing editor at U.S. News. “Our rankings acknowledge these hospitals for their comprehensive excellence, helping families find the very best care conveniently located within their state and community.”
The bulk of the score for each specialty service is based on quality and outcomes data. The process includes a survey of relevant specialists across the country, who are asked to list hospitals they believe provide the best care for patients with the most complex conditions.
The Children’s National specialty services that U.S. News ranked in the top 10 nationally are:
https://innovationdistrict.childrensnational.org/wp-content/uploads/2025/10/USNWR_CNBadges_Set2SideBySide_2025-26-CNRI.jpg385685Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2025/09/InnovationDistrict_CN_WebHeader-1396px-1030x151.pngInnovation District2025-10-07 01:00:072026-01-05 17:33:17Children’s National Hospital once again ranked among the nation’s best by U.S. News & World Report
This November, doctors, fellows and faculty from the Division of Nephrology at Children’s National Hospital will join more than 12,000 kidney professionals at Kidney Week 2025 in Houston, Texas. Hosted annually by the American Society of Nephrology (ASN), this event offers a unique opportunity for attendees to engage in discussions and collaborations focused on the latest breakthroughs and innovations in nephrology.
Professionals and leaders from Children’s National will play a prominent role throughout the weekend, showcasing their expertise in a variety of sessions and presentations including:
Session Moderators
Marva Moxey-Mims, MD: “APOL1-Mediated Kidney Disease: A Blueprint for Precision Nephrology?”
Ashley Trinh, MD (Nephrology Fellow): “Granulomatous Tubulointerstitial Nephritis in Pediatric Inflammatory Bowel Disease: A Case Report”
Jordy Salcedo-Giraldo, MD (Nephrology Fellow): “A Rare Case of Rapidly Progressive Kidney Failure in Alport Syndrome with Crescents”
Jordy Salcedo-Giraldo, Krista Wink, Nicholas Dadzie, Andrew Freiman, MS, CGC & Ashima Gulati, MD, PhD: “Variants of Uncertain Significance Burden Quantification and Reclassification in a Pediatric Kidney Genetics Clinic”
Krista Wink, Jordy Salcedo-Giraldo & Ashima Gulati: “Multidisciplinary Care Needs in Children with ADPKD”
Krista Wink, Jordy Salcedo-Giraldo, Andrew Freiman, Tucker Pyle, MD, PhD & Ashima Gulati: “The Inherited and Polycystic Kidney Disease Program at Children’s National Hospital”
Denver Brown, MD, a pediatric nephrologist at Children’s National Hospital, has been named a 2025 KidneyCure Research Scholar Grant recipient. Her project, titled Metabolic Acidosis and Growth in a Heterogeneous Pediatric CKD Population: A Target Trial Emulation Study, addresses a critical gap in managing chronic kidney disease (CKD) in children.
KidneyCure, established by the American Society of Nephrology, is the largest private funder of kidney-focused research, awarding $3.2 million this year to support 45 investigators. Dr. Brown’s award falls under the Transition to Independence Grants Program, which supports early-career researchers in establishing independent research careers.
Dr. Brown’s study will examine how treating metabolic acidosis affects linear growth in children with CKD, a population in which up to 35% experience growth impairment. “My research focuses on adding to, and increasing, evidence-based approaches to managing chronic kidney disease in children, with a particular focus on metabolic acidosis,” she said.
Instead of a traditional clinical trial, she will use target trial emulation, a method that applies advanced statistical techniques to real-world data from 11 institutions. “Target trial emulation is especially valuable in pediatric nephrology, where the rarity of CKD makes large-scale trials difficult to conduct,” she explained.
Dr. Brown hopes her findings will help reduce morbidity in pediatric CKD patients by identifying effective treatment strategies. “Poor linear growth affects up to 35% of the pediatric CKD population and has been associated with poor health and quality of life outcomes,” she noted.
Looking ahead, she aims to become a leader in pediatric clinical trials. “With the support of this award, I hope to gain skills in advanced statistical approaches that can be leveraged to conduct novel pediatric CKD-focused trials and studies.”
She also brings a personal connection to her work: “My oldest brother is a living-donor kidney transplant recipient. He and my younger brother, the donor, are thriving 15 years post-transplant.”
Her work reflects KidneyCure’s mission to support the next generation of nephrology leaders and drive research that improves patient care.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2025/08/KidneyCure-feature.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2025/09/InnovationDistrict_CN_WebHeader-1396px-1030x151.pngInnovation District2025-08-21 11:26:372026-01-05 17:13:06Denver Brown, MD, awarded prestigious KidneyCure grant to advance pediatric CKD research
Children’s National Hospital hosted its fifteenth annual Research, Education and Innovation Week from March 31–April 4, 2025, bringing together clinicians, scientists, educators and innovators from across the institution to celebrate discovery and collaboration. This year’s theme, “Empowering the Future in Pediatric Research and Innovation with Equity, Technology and a Global Reach,” served as a call to action for advancing science that improves child health both locally and around the world.
Each day of the week-long event featured thought-provoking lectures — now available to watch — dynamic panel discussions, interactive workshops and vibrant poster sessions, all highlighting the diverse and interdisciplinary work taking place across Children’s National.
Centering the patient and the planet
REI Week began on Monday with a powerful keynote lecture from Lynn R. Goldman, MD, MS, MPH, Michael and Lori Milken dean of the Milken Institute School of Public Health at the George Washington University. In her talk, “Children: Uniquely vulnerable to climate-related threats,” Dr. Goldman underscored the urgent need to protect children from the environmental hazards of a changing climate and to integrate climate science into pediatric care and advocacy.
At mid-morning, Mary-Anne “Annie” Hartley, MD, PhD, MPH, director of the LiGHT Laboratory at École Polytechnique Fédérale de Lausanne, introduced the “MOOVE” platform — Massive Open Online Validation and Evaluation of clinical LLMs. Her talk demonstrated how artificial intelligence, when rigorously validated, has the potential to transform clinical decision-making and global health equity.
Monday’s final keynote, “Zinc and childhood diarrhea,” was presented by Christopher Duggan, MD, MPH, director of the Division of Nutrition at Harvard Medical School. Dr. Duggan highlighted the global health impact of zinc supplementation in reducing childhood mortality — a reminder that simple, evidence-based interventions can save millions of lives.
In that first day, the first poster session of the week showcased projects in adolescent medicine, global health, infectious diseases, oncology and more. The session reflected the full breadth of research taking place across Children’s National.
Ambroise Wonkam, MD, PhD, professor of genetic medicine at Johns Hopkins University, then delivered Tuesday’s Global Health Keynote Lecture, “Harnessing our common African genomes to improve health and equity globally.” His work affirmed that inclusive genomics is key to building a healthier world.
Later, the Global Health Initiative event and GCAF Faculty Seminar encouraged attendees to pursue collaborative opportunities at home and abroad, reflecting the growing global footprint of Children’s National research programs.
Transforming education and care delivery
On Wednesday, Larrie Greenberg, MD, professor emeritus of pediatrics, kicked off the day with a Grand Rounds keynote on educational transformation: “Shouldn’t teachers be more collaborative with their learners?” He followed with a CAPE workshop exploring the effectiveness of case-based learning.
In the Jill Joseph Grand Rounds Lecture, Deena J. Chisolm, PhD, director of the Center for Child Health Equity at Nationwide Children’s Hospital, challenged attendees to move beyond dialogue into action in her talk, “Health equity: A scream to a whisper?,” reminding researchers and clinicians that advocacy and equity must be foundational to care.
The day continued with a poster session spotlighting medical education, neonatology, urology and neuroscience, among other fields.
Posters and pathways to progress
Throughout the week, poster sessions highlighted cutting-edge work across dozens of pediatric disciplines. These sessions gave attendees the opportunity to engage directly with investigators and reflect on the shared mission of discovery across multiple disciplines, including:
The REI Week 2025 Awards Ceremony celebrated outstanding contributions in research, mentorship, education and innovation. The winners in each category were:
POSTER SESSION AWARDS
Basic & Translational Research
Faculty: Benjamin Liu, PhD
“Genetic Conservation and Diversity of SARS-CoV-2 Envelope Gene Across Variants of Concern”
Faculty: Steve Hui, PhD
“Brain Metabolites in Neonates of Mothers with COVID-19 Infection During Pregnancy”
Faculty: Raj Shekhar, PhD
“StrepApp: Deep Learning-Based Identification of Group A Streptococcal (GAS) Pharyngitis”
Post docs/Fellows/Residents: Dae-young Kim, PhD
“mhGPT: A Lightweight Domain-Specific Language Model for Mental Health Analysis”
Post docs/Fellows/Residents: Leandros Boukas, MD, PhD
“De Novo Variant Identification From Duo Long-Read Sequencing: Improving Equitable Variant Interpretation for Diverse Family Structures”
Staff: Naseem Maghzian
“Adoptive T Lymphocyte Administration for Chronic Norovirus Treatment in Immunocompromised Hosts (ATLANTIC)”
Graduate Students: Abigail Haffey
“Synergistic Integration of TCR and CAR T Cell Platforms for Enhanced Adoptive Immunotherapy in Brain Tumors”
High School/Undergraduate Students: Medha Pappula
“An ADHD Diagnostic Interface Based on EEG Spectrograms and Deep Learning Techniques”
Clinical Research
Faculty: Folasade Ogunlesi, MD
“Poor Air Quality in Sub-Saharan Africa is Associated with Increase Health Care Utilization for Pain in Sickle Cell Disease Patients”
Faculty: Ayman Saleh, MD
“Growth Parameters and Treatment Approaches in Pediatric ADHD: Examining Differences Across Race”
Post docs/Fellows/Residents: Nicholas Dimenstein, MD, MPH
“Pre-Exposure Prophylaxis (PrEP) Eligibility in the Pediatric Emergency Department”
Staff: Tayla Smith, MPH
“The Public Health Impact of State-Level Abortion and Firearm Laws on Health Outcomes”
Graduate Students: Natalie Ewing
“Patterns of Bacteriuria and Antimicrobial Resistance in Patients Presenting for Primary Cloacal Repair: Is Assisted Bladder Emptying Associated with Bacteriuria?”
Graduate Students: Manuela Iglesias, MS
“Exploring the Relationship Between Child Opportunity Index and Bayley-III Scores in Young Children”
High School/Undergraduate Students: Nicholas Lohman
“Preliminary Findings: The Efficacy, Feasibility and Acceptability of Group Videoconference Cognitive Behavioral Therapy with Exposure and Response Prevention for Treating Obsessive-Compulsive Disorder Among Children and Young People”
Community-Based Research
Faculty: Sharon Shih, PhD “Assessing Pediatric Behavioral Health Access in DC using Secret Shopper Methodology”
Post docs/Fellows/Residents: Georgios Sanidas, MD “Arrested Neuronal Maturation and Development in the Cerebellum of Preterm Infants”
Staff: Sanam Parwani
“Intersectionality of Gender and Sexuality Diversity in Autistic and Non-Autistic Individuals”
Graduate Student: Margaret Dearey “Assessing the Burden of Period Poverty for Youth and Adolescents in Washington, DC: A Pilot Study”
Quality and Performance Improvement
Faculty: Nichole L. McCollum, MD
“A Quality Improvement Study to Increase Nurse Initiated Care from Triage and Improve Timeliness to Care”
Post docs/Fellows/Residents: Hannah Rodriguez, MD
“Reducing Unnecessary Antibiotic Use in a Level IV NICU”
Staff: Amber K. Shojaie, OTD, OTR/L
“Implementing Dynamic Axilla Splints in a Large Burn Patient”
Meleah Boyle, PhD, MPH
“Understanding and Addressing Environmental Sustainability to Protect the Health of the Children’s National and Global Communities”
Eiman Abdulrahman, MD
“Research Capacity Building to Improve Pediatric Emergency and Critical Care in Ethiopia”
Pilot Awards
Alexander Andrews, MD
“EEG as a Diagnostic and Prognostic Marker in Severe Pediatric Malaria, Blantyre Malawi”
Daniel Donoho, MD & Timothy Singer, MD
“Feasibility Study of a Novel Artificial Intelligence-Based Educational Platform to Improve Neurosurgical Operative Skills in Tanzania”
Hasan Syed, MD
“Bridging the Gap an Educational Needs Assessment for Pediatric Neurosurgery Training in Pakistan”
Sofia Perazzo, MD & Lamia Soghier, MD, MEd, MBA
“QI Mentorship to Improve Pediatric Screening and Follow-up in Rural Argentina”
Benjamin Liu, PhD
“AI-Empowered Real-Time Sequencing Assay for Rapid Detection of Schistosomiasis in Senegal”
Rae Mittal, MD
“Assessment and Enhancement of Proficiency in Emergency Child Neurology Topics for Post-Graduate Emergency Medicine Trainees in India”
Innovation Day ignites bold thinking
Thursday, REI Week shifted to the Children’s National Research & Innovation Campus for Innovation Day, a celebration of how bold ideas and collaborative culture can accelerate progress in pediatric medicine.
REI Week 2025 reaffirmed the values that define Children’s National: a commitment to excellence, collaboration and equity in pediatric research and care. As discoveries continue to emerge from our hospital and our research campuses, the connections built and ideas sparked during this week will help shape the future of pediatric health — locally and globally.
By elevating voices from the bedside to the bench, with the support of the executive sponsors Nathan Kuppermann, MD, MBChB, Catherine Bollard, MBChB, MD, Kerstin Hildebrandt, MSHS, Linda Talley, MS, RN, NE-BC and David Wessel, MD, REI Week demonstrated that we must embrace the community in all aspects of our work. Because we know that there are answers we can only get from the patients that we serve—and we need to be their voice.
Research, Education & Innovation Week will be back next year on April 13-17, 2026.
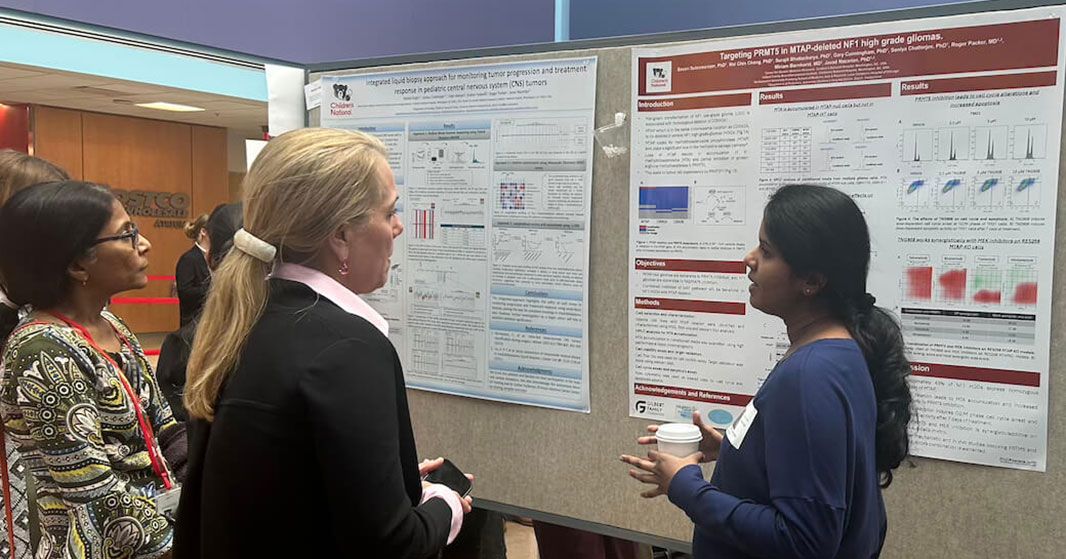
Posters at the REI Week 2025 Monday, March 31 poster session.
Panelists discuss innovation during REI Week 2025.
Global Health Initiative community engagement event during REI Week 2025.
Chris Rees presents his REI Week 2025 lecture.
Nathan Kuppermann listens to a presenter during the REI Week 2025 Tuesday, April 1, poster session.
Michelle Riley-Brown, Nathan Kuppermann, Catherine Bollard and Naomi Luban on stage during the REI Week 2025 awards ceremony.
Brandy Salmon presents on innovation programs at Virginia Tech during the REI Week 2025 Innovation Day.
Catherine Bollard listens to a presenter during the REI Week 2025 Monday, March 21 poster session.
Ambroise Wonkman poses for a picture with Children’s National staff.
Tanzeem Choudhury presenting during REI Week 2025.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2025/04/REI-Week-2025-Monday-Poster-Session-CNRI.jpg385685Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2025/09/InnovationDistrict_CN_WebHeader-1396px-1030x151.pngInnovation District2025-04-22 10:31:052026-04-13 10:39:42REI Week 2025 empowers the future in pediatric research and innovation
The Inherited & Polycystic Kidney Disease (IPKD) Program within the Division of Nephrology at Children’s National Hospital has earned the prestigious designation of Pediatric Center of Excellence by the Polycystic Kidney Disease (PKD) Foundation, the only U.S. organization focused solely on advancing treatments and finding a cure for PKD.
This elite recognition is granted to nephrology practices and clinics that meet the PKD Foundation’s rigorous, patient-centered criteria for specialized PKD care. As a designated Pediatric Center of Excellence, the IPKD Program at Children’s National is recognized for its extensive experience in treating children with PKD and providing multidisciplinary care to meet the unique needs of this patient population. Awarded to only four pediatric hospitals nationwide, this highly selective honor underscores the program’s unwavering commitment to excellence in both patient care and PKD research.
“This achievement is a result of the strong partnership between the PKD Foundation and our Division of Nephrology, working together to elevate the standard of PKD care for our patients and their families,” says Ashima Gulati, MD, PhD, Pediatric Nephrologist and Director of the IPKD Program at Children’s National.
“As providers in the PKD community, we are reminded that our work is part of a broader network, with our patients and caregivers at the heart of everything we do. I’m thrilled that the Division of Nephrology at Children’s National is playing a key role in this initiative, which aligns with the PKD Foundation’s mission to advance promising therapies into clinical practice while ensuring the best possible care for PKD patients today,” Dr. Gulati added.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2025/02/PKDF_PediatricCOEBadge_FY25-Feature.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2025/09/InnovationDistrict_CN_WebHeader-1396px-1030x151.pngInnovation District2025-02-18 12:24:092026-01-05 17:32:47Children’s National designated as Pediatric PKD Center of Excellence
Denver D. Brown, MD, nephrologist at Children’s National Hospital, presented at Kidney Week 2024 on the connection between metabolic acidosis and cardiovascular disease risk in children with chronic kidney disease (CKD).
Denver D. Brown, MD, nephrologist at Children’s National Hospital, presented at Kidney Week 2024 on the connection between metabolic acidosis and cardiovascular disease risk in children with chronic kidney disease (CKD). This research aimed to investigate whether untreated metabolic acidosis could potentially contribute to cardiovascular outcomes in children with CKD, a group at high risk for cardiovascular disease and death from cardiovascular complications. Dr. Brown explains her motivation, findings and future directions for this critical research.
Q: Why did you choose to research this topic?
A: My interest in this area stems from my research interest in pediatric CKD outcomes, with a specific focus on the consequences of metabolic acidosis as well as my larger passion which is to improve the quality and longevity of life for children with CKD. There are several adult focused studies investigating theorized links between untreated metabolic acidosis and poor outcomes such as CKD progression, worse bone health/growth and higher cardiovascular risk. However, in the pediatric CKD population, the consequences of chronic metabolic acidosis remain largely unexplored but is of importance since data shows that nearly one third of children with metabolic acidosis are not receiving treatment to correct their acidosis.
Cardiovascular disease is the number one cause of death in children with CKD. Even though overt cardiovascular disease often does not manifest until adulthood, we do see risk factors emerging during childhood such as high blood pressure and abnormal cholesterol levels. So, if metabolic acidosis contributes to cardiovascular disease risk, it’s crucial to identify and treat it — especially since metabolic acidosis is very treatable
This research was in collaboration with the Chronic Kidney Disease in Children (CKiD) study, which is the largest cohort study of pediatric CKD across North America. It provided robust data on laboratory values, blood pressure, cardiovascular measurements and echocardiograms which allowed me to look deeper into the potential cardiovascular implications of metabolic acidosis.
Q: Where do you see this research going?
A: My ultimate goal is to conduct clinical trials focused on the pediatric CKD population. Children are not well represented in CKD trials as pediatric CKD is relatively rare, making recruitment for studies challenging. However, we can’t keep applying adult data to children because the causes and manifestations of CKD in kids are different.
It is my hope that this research serves as evidence that can be used for a pediatric clinical trial that investigates the benefits of alkali therapy, the treatment for metabolic acidosis. I envision studying how alkali therapy impacts not only cardiovascular outcomes but also growth and other important health domains. The data gathered from my study could ultimately be used in a multi-site trial, aiming to test alkali therapy on a broader scale across various pediatric CKD centers.
Q: Is any work being done between nephrologists and cardiologists to address this issue?
A: This specific study was more exploratory. In adults, the data on the link between metabolic acidosis and cardiovascular disease is mixed. Some studies suggest a connection, while others find no such effect when treating metabolic acidosis. My current work is focused on gathering data to determine if there’s a potential link in the pediatric population and whether it should be examined further in a future trial.
I haven’t collaborated directly with cardiologists on this research. However, this could lead to collaboration with cardiologists down the line.
Q: How is Children’s National Hospital leading the way in this research?
A: Although my research data wasn’t exclusively from Children’s National, the hospital played a pivotal role in the CKiD study, both as an enrollment site and through the involvement of Marva Moxey-Mims, M.D., chief of Nephrology at Children’s National, who played a key role in the CKiD study design and initiation.
Children’s National actively participates in, and encourages, novel and innovative research studies. Being at an institution that prioritizes and contributes to research that advances the health of children has been instrumental in my research career.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2021/10/Denver-Brown.png300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2025/09/InnovationDistrict_CN_WebHeader-1396px-1030x151.pngInnovation District2025-01-29 09:45:292025-01-29 09:47:25The link between metabolic acidosis and cardiovascular disease in children with CKD
Researchers at Children’s National Hospital are developing supervised autonomous robotic surgery to make expert kidney tumor removal accessible in rural areas, combining robotics, AI and surgeon oversight for safer, more precise outcomes.
Imagine a robot capable of planning and executing the intricate removal of a cancerous kidney tumor — a concept that might sound like science fiction. Yet this groundbreaking work is underway at the Sheikh Zayed Institute (SZI) for Pediatric Surgical Innovation at Children’s National Hospital.
Called Supervised Autonomous Robotic Renal Tumor Surgery (SARRTS), the project aims to prove that a supervised autonomous kidney resection is feasible. Its goal is to enable general surgeons in rural hospitals to oversee robots performing complex resections, democratizing access to specialized surgical care. Backed by a $1 million contract from the Advanced Research Projects Agency for Health (ARPA-H), the initiative represents new opportunities in medical innovation.
“The hope is that, someday, patients will no longer have to travel to major oncology centers to get the best possible surgical outcome when faced with renal tumors,” said Kevin Cleary, PhD, associate director of engineering at SZI. “We hope to combine the precision of robotics with a surgeon’s clinical expertise to create consistently high outcomes.”
The patient benefit
Surgery is a cornerstone of cancer treatment, but access to skilled surgeons remains unevenly distributed nationwide. Autonomous robotic surgery could address this disparity by increasing access to expert-level care, enhancing the precision and consistency of procedures and unlocking new surgical possibilities beyond human surgeons’ capabilities.
Under the initial concept, the SARRTS system will use a combination of CT imaging and 3D mapping from a robot’s RGB-depth camera. While the robot independently plans and executes the incision and tumor resection, the supervising surgeon retains full control, with the ability to approve, modify or halt the procedure at any time — an interplay between human expertise and robotic precision to help ensure safety.
Testing will be conducted on realistic kidney models, called phantoms, which are designed to train and test surgical outcomes. The project aims to validate the feasibility of supervised autonomous tumor resection while advancing technologies that could pave the way for broader applications.
“Robotics and medicine have finally reached a point where we can consider projects requiring this level of complexity,” said Anthony Sandler, MD, senior vice president and surgeon-in-chief at Children’s National and executive director of SZI. By combining autonomous robotics, artificial intelligence and surgical expertise, we can profoundly impact the lives of patients facing life-altering cancer diagnoses.”
Children’s National leads the way
In addition to the kidney surgery initiative, the Children’s National team is pursuing other groundbreaking projects. These include a second ARPA-H contract focused on robotic gallbladder removal and a National Institutes of Health grant to explore robotic hip-pinning, a procedure used to repair fractured hips with pins, screws and plates.
Axel Krieger, PhD, an associate professor of mechanical engineering at Johns Hopkins University, is collaborating closely on the kidney resection and gallbladder projects. The interdisciplinary team believes this state-of-the-art care could be tested and developed within the next decade.
“This particular surgery is complex, and a robot may offer advantages to address difficulties created by patient anatomy and visibility within the surgical field,” said Dr. Sandler. “We can imagine a day – in the not too distant future – when a human and a robotic arm could team up to successfully advance this care.”
This project has been funded in whole with federal funds from ARPA-H under cooperative agreement AY1AX000023.
This textbook delves into the psychosocial effects of kidney disease and treatments for children. With chapters written by multidisciplinary experts – including psychologists, nephrologists, neuropsychologists, dietitians, pharmacists, nurses, social workers, child life specialists, as well as patients and families – this book provides a unique and comprehensive perspective of caring for patients with kidney diseases.
The book emphasizes the importance of a multidisciplinary treatment approach, one that incorporates psychosocial factors to ensure the holistic well-being of young patients. It covers a wide range of topics, from disease-specific issues like nutrition and dialysis to broader challenges, such as collaboration with schools, supporting families, advocacy and the transition from pediatric to adult healthcare.
Providing valuable insights into the complexities of managing pediatric kidney diseases, this textbook offers practical strategies for supporting patients throughout their journey, making it an invaluable resource for nephrologists, psychosocial providers, patients and families.
Psychosocial Considerations in Pediatric Kidney Conditions, 1st edition textbook can be purchased here.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2025/01/Psychosocial-Considerations-in-Pediatric-Kidney-Conditions-Book-Cover-feature.jpg466300Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2025/09/InnovationDistrict_CN_WebHeader-1396px-1030x151.pngInnovation District2025-01-08 11:24:012025-01-08 11:26:40Textbook explores psychosocial impacts of kidney disease and provides valuable resources





 Children born with cloacal malformations face many medical challenges beyond surgery. One major concern is kidney health, which may not always be obvious at first. In a recent study published in the
Children born with cloacal malformations face many medical challenges beyond surgery. One major concern is kidney health, which may not always be obvious at first. In a recent study published in the 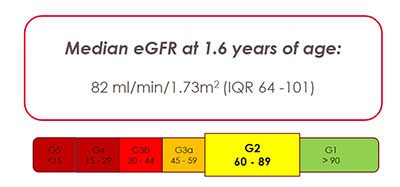








 Children’s National Hospital in Washington, D.C., was ranked as a top hospital in the nation by the U.S. News & World Report 2025-26 Best Children’s Hospitals annual rankings. This marks the ninth straight year Children’s National has made the Honor Roll list. The Honor Roll is a distinction awarded to only 10 children’s hospitals nationwide.
Children’s National Hospital in Washington, D.C., was ranked as a top hospital in the nation by the U.S. News & World Report 2025-26 Best Children’s Hospitals annual rankings. This marks the ninth straight year Children’s National has made the Honor Roll list. The Honor Roll is a distinction awarded to only 10 children’s hospitals nationwide.
 This November, doctors, fellows and faculty from the
This November, doctors, fellows and faculty from the 












 The
The