The best of 2023 from Innovation District
 Advanced MRI visualization techniques to follow blood flow in the hearts of cardiac patients. Gene therapy for pediatric patients with Duchenne muscular dystrophy. 3D-printed casts for treating clubfoot. These were among the most popular articles we published on Innovation District in 2023. Read on for our full list.
Advanced MRI visualization techniques to follow blood flow in the hearts of cardiac patients. Gene therapy for pediatric patients with Duchenne muscular dystrophy. 3D-printed casts for treating clubfoot. These were among the most popular articles we published on Innovation District in 2023. Read on for our full list.
1. Advanced MRI hopes to improve outcomes for Fontan cardiac patients
Cardiac imaging specialists and cardiac surgeons at Children’s National Hospital are applying advanced magnetic resonance imaging visualization techniques to understand the intricacies of blood flow within the heart chambers of children with single ventricle heart defects like hypoplastic left heart syndrome. The data allows surgeons to make critical corrections to the atrioventricular valve before a child undergoes the single ventricle procedure known as the Fontan.
(3 min. read)
2. Children’s National gives first commercial dose of new FDA-approved gene therapy for Duchenne muscular dystrophy
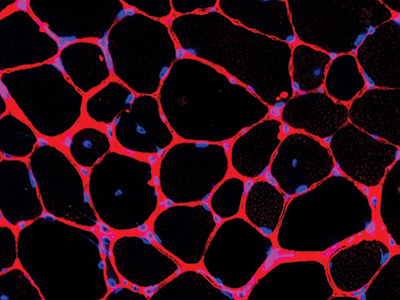
Children’s National Hospital became the first pediatric hospital to administer a commercial dose of Elevidys (delandistrogene moxeparvovec-rokl), the first gene therapy for the treatment of pediatric patients with Duchenne muscular dystrophy (DMD). Elevidys is a one-time intravenous gene therapy that aims to delay or halt the progression of DMD by delivering a modified, functional version of dystrophin to muscle cells.
(2 min. read)
3. New model to treat Becker Muscular Dystrophy
Researchers at Children’s National Hospital developed a pre-clinical model to test drugs and therapies for Becker Muscular Dystrophy (BMD), a debilitating neuromuscular disease that is growing in numbers and lacks treatment options. The work provides scientists with a much-needed method to identify, develop and de-risk drugs for patients with BMD.
(2 min. read)
4. First infants in the U.S. with specially modified pacemakers show excellent early outcomes
In 2022, five newborns with life-threatening congenital heart disease affecting their heart rhythms were the first in the United States to receive a novel modified pacemaker generator to stabilize their heart rhythms within days of birth. Two of the five cases were cared for at Children’s National Hospital. In a follow-up article, the team at Children’s National shared that “early post-operative performance of this device has been excellent.”
(2 min. read)
5. AI: The “single greatest tool” for improving access to pediatric healthcare
Experts from the Food and Drug Administration, Pfizer, Oracle Health, NVIDIA, AWS Health and elsewhere came together to discuss how pediatric specialties can use AI to provide medical care to kids more efficiently, more quickly and more effectively at the inaugural symposium on AI in Pediatric Health and Rare Diseases, hosted by Children’s National Hospital and the Fralin Biomedical Research Institute at Virginia Tech.
(3 min. read)
6. AAP names Children’s National gun violence study one of the most influential articles ever published
The American Academy of Pediatrics (AAP) named a 2019 study led by clinician-researchers at Children’s National Hospital one of the 12 most influential Pediatric Emergency Medicine articles ever published in the journal Pediatrics. The findings showed that states with stricter gun laws and laws requiring universal background checks for gun purchases had lower firearm-related pediatric mortality rates but that more investigation was needed to better understand the impact of firearm legislation on pediatric mortality.
(2 min. read)
7. Why a colorectal transition program matters
Children’s National Hospital recently welcomed pediatric and adult colorectal surgeon Erin Teeple, M.D., to the Division of Colorectal and Pelvic Reconstruction. Dr. Teeple is the only person in the United States who is board-certified as both a pediatric surgeon and adult colorectal surgeon, uniquely positioning her to care for people with both acquired and congenital colorectal disease and help them transition from pediatric care to adult caregivers.
(3 min. read)
8. First-of-its-kind holistic program for managing pain in sickle cell disease
The sickle cell team at Children’s National Hospital received a grant from the Founders Auxiliary Board to launch a first-of-its-kind, personalized holistic transformative program for the management of pain in sickle cell disease. The clinic uses an inter-disciplinary approach of hematology, psychology, psychiatry, anesthesiology/pain medicine, acupuncture, mindfulness, relaxation and aromatherapy services.
(3 min read)
9. Recommendations for management of positive monosomy X on cell-free DNA screening
Non-invasive prenatal testing using cell-free DNA (cfDNA) is currently offered to all pregnant women regardless of the fetal risk. In a study published in the American Journal of Obstetrics and Gynecology, researchers from Children’s National Hospital provided context and expert recommendations for maternal and fetal evaluation and management when cfDNA screening is positive for monosomy X or Turner Syndrome.
(2 min. read)
10. Innovation in clubfoot management using 3D anatomical mapping
While clubfoot is relatively common and the treatment is highly successful, the weekly visits required for Ponseti casting can be a significant burden on families. Researchers at Children’s National Hospital are looking for a way to relieve that burden with a new study that could eliminate the weekly visits with a series of 3D-printed casts that families can switch out at home.
(1 min. read)
11. Gender Self-Report seeks to capture the gender spectrum for broad research applications
A new validated self-report tool provides researchers with a way to characterize the gender of research participants beyond their binary designated sex at birth. The multi-dimensional Gender Self-Report, developed using a community-driven approach and then scientifically validated, was outlined in a peer-reviewed article in the American Psychologist, a journal of the American Psychological Association.
(2 min. read)
12. Cardiovascular and bone diseases in chronic kidney disease
In a study published by Advances in Chronic Kidney Disease, a team at Children’s National Hospital reviewed cardiovascular and bone diseases in chronic kidney disease and end-stage kidney disease patients with a focus on pediatric issues and concerns.
(1 min. read)


 Unraveling how cells mend after injury serves as a key to unlocking potential therapies. Recent findings from the
Unraveling how cells mend after injury serves as a key to unlocking potential therapies. Recent findings from the 



















 Children’s National Hospital named
Children’s National Hospital named 








 Children’s National Hospital in Washington, D.C., was ranked #5 in the nation on the U.S. News & World Report 2023-24 Best Children’s Hospitals annual rankings. This marks the seventh straight year Children’s National has made the Honor Roll list. The Honor Roll is a distinction awarded to only 10 children’s hospitals nationwide.
Children’s National Hospital in Washington, D.C., was ranked #5 in the nation on the U.S. News & World Report 2023-24 Best Children’s Hospitals annual rankings. This marks the seventh straight year Children’s National has made the Honor Roll list. The Honor Roll is a distinction awarded to only 10 children’s hospitals nationwide.