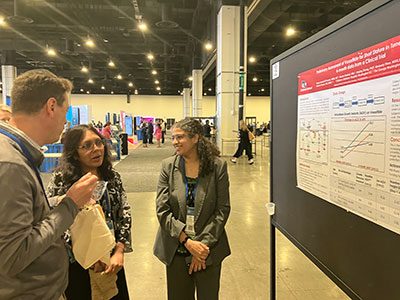
Roopa Shankar, MD, an endocrinologist at Children’s National Hospital and principal investigator on the study, presented these findings at the Pediatric Endocrine Society annual meeting.
In the world’s first clinical trial of vosoritide for Turner Syndrome (TS), preliminary results show an increase in annualized growth velocity (AGV) in girls with TS in the first six-months of the trial.
Roopa Shankar, MD, an endocrinologist at Children’s National Hospital and principal investigator on the study, presented these findings at the Pediatric Endocrine Society annual meeting on May 17.
The big picture
TS is a rare genetic disorder that occurs in one to about 2,500 girls and is caused by a partial or complete missing X chromosome. Some of the characteristics of TS are short stature, delayed puberty, kidney, thyroid and heart problems. Although there is no cure for TS, many of the symptoms can be treated.
Previously approved for use in achondroplasia, vosoritide is a C-type natriuretic peptide (CNP) analog that binds its receptor on healthy cartilage cells called chondrocytes, leading to increased chondrocyte proliferation and differentiation via its inhibition of the ERK1/2-MAPK pathway.
To date, four participants are enrolled in the trial, and three have completed six months of treatment. Participants with prior growth hormone (GH) exposure increased AGV by +4.9 and +1.57 cm/year over the baseline AGV on GH treatment. The participant without prior GH exposure had an increased AGV +6.04 cm above the baseline at the six-month visit.
What’s next
“The increase in AGV with vosoritide is comparable to the response expected with GH treatment in the GH naïve patient,” says Dr. Shankar. “What’s particularly encouraging is the higher AGV seen in patients previously treated with GH — compared to their baseline on GH alone—which suggests vosoritide may offer additional growth benefits, even when GH responsiveness has diminished. While these preliminary results are very promising, we need longer-term data to establish the safety and the sustained impact on growth. Recruitment for this study is ongoing.”
Additional authors from Children’s National include: Niusha Shafaei, MSc, Anqing Zhang, PhD, Kimberly Pitner, MSHS, BSN, RN, Niti Dham, MD, Andrew Dauber, MD, MMSc.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2025/05/poster-presentation-at-PES-feature.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2025-05-23 13:20:272025-05-23 13:21:22Encouraging early results for vosoritide in Turner syndrome
Dr. Tarini has extensively studied policies to optimize the delivery of genetic services for newborns and their families.
Children’s National Hospital named Beth A. Tarini, MD, MS, MBA, as the Richard and Agnes Hudson Endowed Chair in Health Services Research at Children’s National Hospital.
Dr. Tarini, a pediatrician, is the Co-Director of the Center for Translational Research in the Children’s National Research Institute, the hospital’s Director of Resident Research and a professor of pediatrics at George Washington University.
The big picture
Dr. Tarini joins a distinguished group of Children’s National physicians and scientists with an endowed chair. Children’s National is grateful to generous donors who have funded 51 professorships altogether.
Professorships support groundbreaking work on behalf of children and their families and foster new discoveries and innovations in pediatric medicine. These appointments carry prestige and honor that reflect the recipient’s achievements and the donor’s commitment to advancing and sustaining knowledge.
Why it matters
Dr. Tarini has extensively studied policies to optimize the delivery of genetic services for newborns and their families. She has obtained $10 million in federal and foundation funding. A national leader in her field, she has served as president of the Society for Pediatric Research and as an appointed member of the Advisory Committee on Heritable Disorders in Newborns and Children. In the latter role, she helped advise the Secretary of the U.S. Department of Health and Human Services on the most appropriate application of universal newborn screening tests, technologies, policies, guidelines and standards.
“It’s an honor to receive the Hudson Chair, which allows me to bridge research and real-world impact,” says Dr. Tarini. “With this support, I will continue working to translate scientific discovery into better genetic services and policy for all newborns and their families.”
The visionary investment from the estate of Richard and Agnes Hudson will ensure that Dr. Tarini and future chairholders can launch bold new initiatives to rapidly advance the field of health services research, elevate the hospital’s academic leadership and improve the health and well-being of children.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2025/05/Beth-Tarini-feature.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2025-05-19 10:54:132025-05-19 10:56:26Honor bestowed on Beth A. Tarini, MD, MS, MBA
Children’s National Hospital hosted its fifteenth annual Research, Education and Innovation Week from March 31–April 4, 2025, bringing together clinicians, scientists, educators and innovators from across the institution to celebrate discovery and collaboration. This year’s theme, “Empowering the Future in Pediatric Research and Innovation with Equity, Technology and a Global Reach,” served as a call to action for advancing science that improves child health both locally and around the world.
Each day of the week-long event featured thought-provoking lectures — now available to watch — dynamic panel discussions, interactive workshops and vibrant poster sessions, all highlighting the diverse and interdisciplinary work taking place across Children’s National.
Centering the patient and the planet
REI Week began on Monday with a powerful keynote lecture from Lynn R. Goldman, MD, MS, MPH, Michael and Lori Milken dean of the Milken Institute School of Public Health at the George Washington University. In her talk, “Children: Uniquely vulnerable to climate-related threats,” Dr. Goldman underscored the urgent need to protect children from the environmental hazards of a changing climate and to integrate climate science into pediatric care and advocacy.
At mid-morning, Mary-Anne “Annie” Hartley, MD, PhD, MPH, director of the LiGHT Laboratory at École Polytechnique Fédérale de Lausanne, introduced the “MOOVE” platform — Massive Open Online Validation and Evaluation of clinical LLMs. Her talk demonstrated how artificial intelligence, when rigorously validated, has the potential to transform clinical decision-making and global health equity.
Monday’s final keynote, “Zinc and childhood diarrhea,” was presented by Christopher Duggan, MD, MPH, director of the Division of Nutrition at Harvard Medical School. Dr. Duggan highlighted the global health impact of zinc supplementation in reducing childhood mortality — a reminder that simple, evidence-based interventions can save millions of lives.
In that first day, the first poster session of the week showcased projects in adolescent medicine, global health, infectious diseases, oncology and more. The session reflected the full breadth of research taking place across Children’s National.
Ambroise Wonkam, MD, PhD, professor of genetic medicine at Johns Hopkins University, then delivered Tuesday’s Global Health Keynote Lecture, “Harnessing our common African genomes to improve health and equity globally.” His work affirmed that inclusive genomics is key to building a healthier world.
Later, the Global Health Initiative event and GCAF Faculty Seminar encouraged attendees to pursue collaborative opportunities at home and abroad, reflecting the growing global footprint of Children’s National research programs.
Transforming education and care delivery
On Wednesday, Larrie Greenberg, MD, professor emeritus of pediatrics, kicked off the day with a Grand Rounds keynote on educational transformation: “Shouldn’t teachers be more collaborative with their learners?” He followed with a CAPE workshop exploring the effectiveness of case-based learning.
In the Jill Joseph Grand Rounds Lecture, Deena J. Chisolm, PhD, director of the Center for Child Health Equity at Nationwide Children’s Hospital, challenged attendees to move beyond dialogue into action in her talk, “Health equity: A scream to a whisper?,” reminding researchers and clinicians that advocacy and equity must be foundational to care.
The day continued with a poster session spotlighting medical education, neonatology, urology and neuroscience, among other fields.
Posters and pathways to progress
Throughout the week, poster sessions highlighted cutting-edge work across dozens of pediatric disciplines. These sessions gave attendees the opportunity to engage directly with investigators and reflect on the shared mission of discovery across multiple disciplines, including:
The REI Week 2025 Awards Ceremony celebrated outstanding contributions in research, mentorship, education and innovation. The winners in each category were:
POSTER SESSION AWARDS
Basic & Translational Research
Faculty: Benjamin Liu, PhD
“Genetic Conservation and Diversity of SARS-CoV-2 Envelope Gene Across Variants of Concern”
Faculty: Steve Hui, PhD
“Brain Metabolites in Neonates of Mothers with COVID-19 Infection During Pregnancy”
Faculty: Raj Shekhar, PhD
“StrepApp: Deep Learning-Based Identification of Group A Streptococcal (GAS) Pharyngitis”
Post docs/Fellows/Residents: Dae-young Kim, PhD
“mhGPT: A Lightweight Domain-Specific Language Model for Mental Health Analysis”
Post docs/Fellows/Residents: Leandros Boukas, MD, PhD
“De Novo Variant Identification From Duo Long-Read Sequencing: Improving Equitable Variant Interpretation for Diverse Family Structures”
Staff: Naseem Maghzian
“Adoptive T Lymphocyte Administration for Chronic Norovirus Treatment in Immunocompromised Hosts (ATLANTIC)”
Graduate Students: Abigail Haffey
“Synergistic Integration of TCR and CAR T Cell Platforms for Enhanced Adoptive Immunotherapy in Brain Tumors”
High School/Undergraduate Students: Medha Pappula
“An ADHD Diagnostic Interface Based on EEG Spectrograms and Deep Learning Techniques”
Clinical Research
Faculty: Folasade Ogunlesi, MD
“Poor Air Quality in Sub-Saharan Africa is Associated with Increase Health Care Utilization for Pain in Sickle Cell Disease Patients”
Faculty: Ayman Saleh, MD
“Growth Parameters and Treatment Approaches in Pediatric ADHD: Examining Differences Across Race”
Post docs/Fellows/Residents: Nicholas Dimenstein, MD, MPH
“Pre-Exposure Prophylaxis (PrEP) Eligibility in the Pediatric Emergency Department”
Staff: Tayla Smith, MPH
“The Public Health Impact of State-Level Abortion and Firearm Laws on Health Outcomes”
Graduate Students: Natalie Ewing
“Patterns of Bacteriuria and Antimicrobial Resistance in Patients Presenting for Primary Cloacal Repair: Is Assisted Bladder Emptying Associated with Bacteriuria?”
Graduate Students: Manuela Iglesias, MS
“Exploring the Relationship Between Child Opportunity Index and Bayley-III Scores in Young Children”
High School/Undergraduate Students: Nicholas Lohman
“Preliminary Findings: The Efficacy, Feasibility and Acceptability of Group Videoconference Cognitive Behavioral Therapy with Exposure and Response Prevention for Treating Obsessive-Compulsive Disorder Among Children and Young People”
Community-Based Research
Faculty: Sharon Shih, PhD “Assessing Pediatric Behavioral Health Access in DC using Secret Shopper Methodology”
Post docs/Fellows/Residents: Georgios Sanidas, MD “Arrested Neuronal Maturation and Development in the Cerebellum of Preterm Infants”
Staff: Sanam Parwani
“Intersectionality of Gender and Sexuality Diversity in Autistic and Non-Autistic Individuals”
Graduate Student: Margaret Dearey “Assessing the Burden of Period Poverty for Youth and Adolescents in Washington, DC: A Pilot Study”
Quality and Performance Improvement
Faculty: Nichole L. McCollum, MD
“A Quality Improvement Study to Increase Nurse Initiated Care from Triage and Improve Timeliness to Care”
Post docs/Fellows/Residents: Hannah Rodriguez, MD
“Reducing Unnecessary Antibiotic Use in a Level IV NICU”
Staff: Amber K. Shojaie, OTD, OTR/L
“Implementing Dynamic Axilla Splints in a Large Burn Patient”
Meleah Boyle, PhD, MPH
“Understanding and Addressing Environmental Sustainability to Protect the Health of the Children’s National and Global Communities”
Eiman Abdulrahman, MD
“Research Capacity Building to Improve Pediatric Emergency and Critical Care in Ethiopia”
Pilot Awards
Alexander Andrews, MD
“EEG as a Diagnostic and Prognostic Marker in Severe Pediatric Malaria, Blantyre Malawi”
Daniel Donoho, MD & Timothy Singer, MD
“Feasibility Study of a Novel Artificial Intelligence-Based Educational Platform to Improve Neurosurgical Operative Skills in Tanzania”
Hasan Syed, MD
“Bridging the Gap an Educational Needs Assessment for Pediatric Neurosurgery Training in Pakistan”
Sofia Perazzo, MD & Lamia Soghier, MD, MEd, MBA
“QI Mentorship to Improve Pediatric Screening and Follow-up in Rural Argentina”
Benjamin Liu, PhD
“AI-Empowered Real-Time Sequencing Assay for Rapid Detection of Schistosomiasis in Senegal”
Rae Mittal, MD
“Assessment and Enhancement of Proficiency in Emergency Child Neurology Topics for Post-Graduate Emergency Medicine Trainees in India”
Innovation Day ignites bold thinking
Thursday, REI Week shifted to the Children’s National Research & Innovation Campus for Innovation Day, a celebration of how bold ideas and collaborative culture can accelerate progress in pediatric medicine.
REI Week 2025 reaffirmed the values that define Children’s National: a commitment to excellence, collaboration and equity in pediatric research and care. As discoveries continue to emerge from our hospital and our research campuses, the connections built and ideas sparked during this week will help shape the future of pediatric health — locally and globally.
By elevating voices from the bedside to the bench, with the support of the executive sponsors Nathan Kuppermann, MD, MBChB, Catherine Bollard, MBChB, MD, Kerstin Hildebrandt, MSHS, Linda Talley, MS, RN, NE-BC and David Wessel, MD, REI Week demonstrated that we must embrace the community in all aspects of our work. Because we know that there are answers we can only get from the patients that we serve—and we need to be their voice.
Research, Education & Innovation Week will be back next year on April 13-17, 2026.
Posters at the REI Week 2025 Monday, March 31 poster session.
Panelists discuss innovation during REI Week 2025.
Global Health Initiative community engagement event during REI Week 2025.
Chris Rees presents his REI Week 2025 lecture.
Nathan Kuppermann listens to a presenter during the REI Week 2025 Tuesday, April 1, poster session.
Michelle Riley-Brown, Nathan Kuppermann, Catherine Bollard and Naomi Luban on stage during the REI Week 2025 awards ceremony.
Brandy Salmon presents on innovation programs at Virginia Tech during the REI Week 2025 Innovation Day.
Catherine Bollard listens to a presenter during the REI Week 2025 Monday, March 21 poster session.
Ambroise Wonkman poses for a picture with Children’s National staff.
Tanzeem Choudhury presenting during REI Week 2025.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2025/04/REI-Week-2025-Monday-Poster-Session-CNRI.jpg385685Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2025-04-22 10:31:052025-06-10 12:20:52REI Week 2025 empowers the future in pediatric research and innovation
Jo Lynne Rokita, PhD, is the director of the new Bioinformatics Core housed within the Brain Tumor Institute at Children’s National Hospital.
Jo Lynne Rokita, PhD, is the director of the new Bioinformatics Core housed within the Brain Tumor Institute at Children’s National Hospital. Dr. Rokita is a cancer genomics leader with 20 years of combined research experience in academia, industry and the government. She’s also a technical and analytical expert in genomics research using microarrays and high-throughput sequencing.
“We are very excited that we were able to recruit Dr. Rokita as director of the Bioinformatics Core Facility,” says Muller Fabbri, MD, PhD, associate center director for Cancer and Immunology Research at Children’s National. “Her Bioinformatics Core will play a central role in providing the Brain Tumor Institute community with unique expertise spanning biology/genetics/genomics and bioinformatics and will propel Children’s National forward as a national and worldwide leader in pediatric brain tumor research.”
Dr. Rokita is overseeing the core’s creation, including bringing both bioinformatics staff and computing infrastructure to the program, which will support the data analysis needs of the institute’s investigators. She recently answered questions about the new core and also talked a little bit about the focus of her own research that will continue at Children’s National.
Q: Why is the Brain Tumor Institute establishing a Bioinformatics Core?
A: Growing the institute’s bioinformatics capabilities was one of the things that leadership wanted to make sure was built into the plan for the record-setting $96 million gift that was received in 2023. There was a clear need among the principal investigators for this type of research support which includes organization, analysis and interpretation of large-scale genetic sequencing and other “-omics” data.
Q: How did you decide to join Children’s National?
A: I was leading a pediatric brain tumor focused bioinformatics team at Children’s Hospital of Philadelphia (CHOP). As a part of the Children’s Brain Tumor Network (CBTN), I worked closely with a collaborator from Children’s National, Brian Rood, MD, medical director of the Brain Tumor Institute. He told me about the opportunity and I was very excited to apply.
Q: How did your previous work prepare you for this role?
A: I’ve spent the past 10 years in the pediatric cancer field with the last six focused on brain tumor research. In my various roles at CHOP, I led multiple large-scale genomic analysis efforts, comprehensive data and methods for which we then provided openly to the community. During my postdoctoral fellowship, these efforts included a large neuroblastoma patient-derived cell line “ENCODE” as well as a resource led in collaboration with multiple institutes and the National Cancer Institute funded by Alex’s Lemonade Stand Foundation (ALSF). We further scaled these efforts to build open analytical platforms to empower researchers to build upon our work doing their own cancer genomic analysis. In collaboration with the Childhood Cancer Data Lab at ALSF, we built the platform that ultimately ballooned into the OpenPedCan includes large amounts of harmonized genomic, epigenomic and proteomic data for patients with pediatric cancer. What’s unique is that the data is all processed in the same way and easily accessible through multiple mechanisms. Researchers can use these data to ask questions about the cancer type they study or genes of interest. For example, genes over-expressed, absent and/or mutated in a specific tumor subtype may lead to a better understanding of how a patient’s cancer may respond to a treatment.
We’ll be bringing some of the workflows we created previously here to Children’s National, and that will allow us to join newly generated internal data with the thousands of data points we’ve already harmonized using these workflows.
Q: Can you give us some examples of how data harmonization benefits the field of pediatric brain tumor research?
A: Harmonizing across institutions and databases will help us increase the number of data points available for study. This is really important for rare types of tumors and are major foci of institute collaborator Adriana Fonseca, MD, and her International Rare Brain Tumor Registry program. The Bioinformatics Core will support data organization and analysis for this effort, which aims to sequence the rarest brain tumors — those that make up between only 3% and 5% of all brain tumors. If all the data is analyzed the same way, we can combine multiple studies to increase our total dataset, which in turn may reveal new biomarkers and new subtypes of those tumors. It is critical that we continue to build these data resources in a way that they can be accessed by everyone doing this work. Having dedicated support systems for these functions will push the research farther, faster.
Q: As this work gets underway, what is the core’s main function?
A: As this initiative gets underway, the Bioinformatics Core’s primary goal is to empower investigators by streamlining and centralizing data analyses. We help researchers transfer sequencing data into secure cloud storage, organize newly generated records and prepare those datasets for in-depth study. Our bioinformatics scientists then perform downstream analyses to address the specific questions posed by each investigator. On the backend, we collaborate with information technology at Children’s National to develop a robust infrastructure that supports these activities efficiently. By offering these services in-house, we aim to ensure our investigators have seamless, comprehensive support—ultimately driving innovation and accelerating research progress.
Q: What is “open science” and why is it important in bioinformatics?
A: One of our big focus areas is open science, meaning our goal is to push data and code out into the community so that researchers can easily reproduce and build upon our findings. I’m excited to bring the principles of open science, code sharing and data sharing to the Bioinformatics Core.
Making resources open makes it easier for teams to work together across institutions and research programs. It is also going to benefit patients because people can reuse the code and move towards cures faster. For example, we try to package an entire manuscript’s code when we provide our data so it’s clear how the analyses were done.
Q: What is your particular research passion?
A: I work in several research areas and with many brilliant collaborators. One of our focus areas is understanding how RNA splicing can contribute to pediatric brain tumors to create a change in a protein. We have recently identified tumor-specific splice events in some pediatric brain tumor types and will be partnering with Dalia Haydar, PharmD, PhD, to create therapeutic approaches to targeting these. We are also developing a user-friendly application for mining the large amount of splicing data in pediatric brain tumors.
Another focus of our lab is understanding how the patient’s host genome (alterations inherent in their blood DNA) influences the tumor’s genetics. For example, we’ve just preprinted a study connecting inherited variants to tumor genetics and patient outcomes.
Finally, we are interested in how differences in race, ethnicity and social determinants of health influence survival and treatment outcomes for children with brain tumors.
I am passionate about data sharing, code reproducibility and promoting open science in general.
Q: Is there any specific reason you decided to focus your work around brain tumors and pediatric brain tumors?
A: My cousin passed away from a brain tumor when I was in high school. They didn’t have molecular diagnosis then, but he had a brainstem glioma, likely a diffuse midline glioma. In graduate school, I studied addiction genetics and became fascinated with the brain and towards the end, cancer. As an alumna of Penn State, I was actively involved in philanthropic events raising money for their Dance MaraTHON supporting children with cancer. I was lucky to land a postdoc at CHOP and lean into subsequent roles which allowed my passion for this field to grow.
Q: Last question — What do you do with your time when you are not studying pediatric brain tumor data?
A: I enjoy being with my family, observing my children learn and grow, and listening to music.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2025/02/bioinformatics-feature.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2025-02-26 16:09:002025-03-06 13:18:03Q&A with Dr. Rokita: Building bioinformatics infrastructure at the Brain Tumor Institute
Children’s National is ranked one of the top 10 pediatric hospitals in the nation by U.S. News & World Report. Our faculty and staff are proud of the impact made on the lives of children and families in our community. Your participation in the U.S. News & World Report annual reputational survey validates the quality of care we provide and reflects the mutual respect and trust we share as healthcare professionals.
How to determine your voting eligibility
Voting for the U.S. News & World Report Best Children’s Hospitals rankings can be done only through Doximity.
To participate, physicians must:
Be board-certified and meet the eligibility criteria for the voting categories.
For child and adolescent psychologists, your account must be up to date with your specialty and subspecialty correctly marked.
Be a credential-verified member of Doximity (you must have an active and claimed Doximity profile).
Have all certifications and board documents currently up-to-date in your Doximity profile.
You have to claim your profile on Doximity.com to participate in the online survey. If you have not yet claimed your Doximity profile, go to Doximity.com, and click “Find My Profile.”
Once your profile has been claimed, you must confirm your email address and board certifications.
Verified Doximity members will receive an email inviting them to participate in the U.S. News survey.
For more information on how to claim your profile, visit Doximity.com
How to update and verify existing Doximity account information
Your Doximity profile must have up-to-date licenses, certifications and board documents.
Once you are logged in, your profile will automatically be in “Edit Mode.” You are able to add new items or edit existing information.
Update your Doximity profile and ensure your information is current.
Once registered, users wishing to participate in the online survey should:
Watch for an email from Doximity about the annual member survey.
Even if you don’t see the email, if you are a registered Doximity user, you can still vote by logging in to Doximity.com with your username and password during the voting period.
Once logged in, look for a U.S. News graphic or button on the homepage and click on it.
The survey asks users to name the hospitals that provide the best care in your respective specialty, without consideration to location or cost. Pediatric specialists will list 10 hospitals. The order in which you list the hospitals does not matter.
Please note: Children’s National Hospital is listed as “Children’s National Hospital Washington, DC” on the survey.
Visit Doximity’s FAQs if you have issues or questions about registration or claiming your profile.
How to cast your vote
In February 2025 when voting opens, all survey-eligible physicians will receive a notification on the Doximity app for Android or iOS. If you do not use the Doximity app, you will receive an email when voting opens.
Log in to your Doximity account at doximity.com or via the mobile app.
Click the Notifications icon or tap the “Submit your Nominations” button on the homepage. You can also search for “U.S. News Best Hospitals”
Select 10 hospitals in your respective specialty that you believe provide the best care in the United States.
Submit your vote
Having technical issues?
If you have difficulty registering with Doximity or completing the survey, please visit Doximity support for assistance.
Vote
The 2025 U.S. News & World Report Best Children’s Hospitals reputation voting will open in mid-February. Look for your Doximity notification to vote.
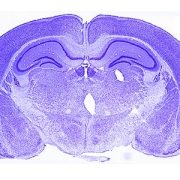
A cross-disciplinary team of researchers and physicians from Children’s National Hospital and Washington University School of Medicine in St. Louis, in collaboration with physicians from around the world, identified a new genetic cause of neurodevelopmental disorders (NDDs). In a new study published in the American Journal of Human Genetics, researchers found 14 unrelated patients with 15 different sequence variants in HECTD1 – 10 missense, 3 frameshift, 1 nonsense and 1 splicing variant – with NDDs, including autism, attention-deficit/hyperactivity disorder (ADHD) and epilepsy.
Moving the field forward
Many patients suspected of having a genetic disorder remain undiagnosed. In about 10% of these cases, the genetic change is in a gene unknown to cause the disorder.
“Describing a new genetic cause of neurodevelopmental disorders will allow for the characterization of the gene’s role in brain development, the genetic syndrome and the mechanisms of disease,” says Irene Zohn, PhD, principal investigator in the Center for Genetic Medicine Research at Children’s National and co-lead of the study. “This information will lead to developing treatments to improve the lives of patients.”
The patient benefit
“Our study represents the first report of HECTD1 in NDDs,” says Dr. Zohn. “Now that this gene is linked to the disorder, clinicians with patients with sequence variation in this gene can enroll in studies to understand this new HECTD1 syndrome.”
Proper genetic diagnosis is important so that comorbidities and the natural history of the disorder can be described, which will lead to improvements in patient care.
What we hope to discover
Now that a new genetic syndrome has been defined, researchers hope to establish how prevalent the syndrome is and describe its features. Using pre-clinical models, they’ll continue to study the developmental basis of the disorder and the molecular mechanisms to develop therapies.
Children’s National leads the way
The HECTD1 gene was discovered in Dr. Zohn’s laboratory, and her research team connected with Christina Gurnett, MD, PhD, co-lead of the study from Washington University School of Medicine, to link this gene to human disease.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2025/01/zohn_brain_picture-feature.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2025-01-28 11:58:372025-06-11 15:38:46Study finds new genetic cause of neurodevelopmental disorders
The Children’s National 2023-2024 Academic Annual Report show on a tablet.
Children’s National Hospital has released its 2023-2024 Academic Annual Report, showcasing a year of transformative progress in pediatric medicine. The report highlights achievements across its research centers, institutes and Innovation Ventures, underscoring the hospital’s role as a leader in advancing child health through innovation and collaboration.
“This year’s report reflects the remarkable progress we have made in advancing the frontiers of pediatric medicine,” said Nathan Kuppermann, MD, MPH, Chief Academic Officer and Chair of Pediatrics. “It highlights groundbreaking work across our research centers, institutes, and Innovation Ventures, showcasing the collaborative spirit that drives our mission forward. These achievements underscore our shared commitment to delivering transformative research and the best possible outcomes for children and families.”
Delivering across centers
The report captures the contributions of each of Children’s National’s research centers, each pushing the boundaries of pediatric healthcare:
Center for Cancer & Immunology Research (CCIR): Delivering on the promise of cell and gene therapies, offering innovative treatments for pediatric cancers and immune disorders.
Center for Genetic Medicine Research (CGMR): Advancing pediatric genetic medicine through interdisciplinary efforts, addressing complex genetic conditions with cutting-edge science.
Center for Neuroscience Research (CNR): A year of growth in scientific excellence, advancing the understanding of brain development and neurological conditions.
Center for Prenatal, Neonatal & Maternal Health Research (CPHNMR): Revolutionizing neonatal care with its pioneering infant brain health neuromonitoring program.
Center for Translational Research (CTR): Facilitating groundbreaking work by new K awardees and driving translational research to bridge the gap between discovery and clinical care.
Sheikh Zayed Institute for Pediatric Surgical Innovation (SZI): Leading the way in advanced research projects in pediatric surgery, pushing technological boundaries to improve outcomes for children worldwide.
Taking the lead in innovation through collaboration
Innovation Ventures at Children’s National is advancing pediatric health security, addressing unique challenges with transformative solutions. Meanwhile, the Children’s National Research & Innovation Campus (CNRIC) continues to thrive as a hub for discovery and collaboration, hosting conferences on topics like artificial intelligence in healthcare, cell and gene therapy, and pediatric epilepsy research.
A vision for the future
The report also highlights Children’s National’s focus on integrating cutting-edge technologies like artificial intelligence into its research and clinical practices, as well as addressing global health challenges such as the effects of climate change on children’s health. These efforts reflect the hospital’s commitment to improving outcomes for children everywhere through innovation, teamwork, and forward-thinking leadership.
The 2023-2024 Academic Annual Report serves as a testament to the dedication and expertise of the Children’s National community, showcasing how collaboration and innovation are shaping the future of pediatric healthcare.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2025/01/2023-2024-Academic-Annual-Report-cover-feature.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2025-01-13 13:51:482025-01-13 13:53:49Children’s National delivers on the promise in 2024
Meet the winners (left to right): Syed M. Anwar, Ph.D., M.S., principal investigator at Children’s National; Daniel Capellan Martin, M.Sc., Polytechnic University of Madrid; Abhijeet Parida, data scientist at Children’s National; and Austin Tapp, Ph.D., postdoctoral research fellow at Children’s National.
Using an award-winning artificial intelligence (AI) algorithm developed at Children’s National Hospital, researchers ranked first in the world in the Brain Tumor Segmentation-Africa (BraTS-Africa) challenge for their approach to identifying different parts of deadly gliomas. The details of their innovative method were recently published on arXiv, a curated research-sharing platform.
“Technology can bridge the gap in healthcare between high- and low-resource countries,” said Marius George Linguraru, D.Phil., M.A., M.Sc., the Connor Family Professor in Research and Innovation and principal investigator in the Sheikh Zayed Institute for Pediatric Surgical Innovation (SZI). “By tailoring methods we created at our hospital to fit the needs of specific regions, such as sub-Saharan Africa, our research helps improve medical imaging and diagnosis in challenging environments.”
Dr. Linguraru was the program chair of the International Conference on Medical Image Computing and Computer Assisted Intervention (MICCAI) 2024 in Marrakesh, Morocco, the leading global meeting on AI in medical imaging.
Children’s National leads the way
Gliomas are a type of brain tumor with a high death rate and are especially difficult to diagnose in low- and middle-income countries. Given the increased need in Africa, researchers worldwide came together in Morocco to compete over the best way to accurately detect and measure tumors using MRI data and AI.
By applying advanced machine-learning techniques, the researchers adapted tools initially designed for well-resourced settings to work in countries with far fewer.
The study focused on transfer learning, a process in which an AI model is trained in advance on a large number of brain tumor images and then adjusted to work with smaller sets of new data. In this case, the models were adapted to work with local sub-Saharan African data using a strategy to combine different models’ strengths.
When tested, the approach achieved impressive accuracy scores. The Children’s National team, which included a colleague from the Polytechnic University of Madrid, ranked first in the BraTS-Africa 2024 challenge for identifying different parts of gliomas.
“To make the method widely available, the winning algorithm is shared online for others to use and improve upon,” Dr. Linguraru said. “My favorite part of these competitions is how they highlight the way innovation and collaboration can reduce global healthcare inequalities.”
The big picture
Children’s National researchers consistently lead global events using AI and advanced imaging to tackle complex healthcare challenges. In 2023, the team won a global contest to measure pediatric brain tumors at the MICCAI 2023 Conference. This year’s success in the BraTS-Africa challenge builds on this knowledge base and expands its use to adult gliomas.
At the Radiological Society of North America 2024 annual meeting, which drew 50,000 attendees, Zhifan Jiang, Ph.D., a staff scientist in the Precision Medical Imaging Lab at SZI, also won the Cum Laude Award for his scientific poster on applying AI to radiological images to predict severe outcomes for children with brain tumors caused by neurofibromatosis type 1.
“These achievements show how our science is leading the world in using AI for good,” Dr. Linguraru said. “Every day, we’re building on our knowledge of advanced imaging, brain tumors and AI to improve the diagnosis, measurement and treatment of deadly tumors — on a global scale.”
https://innovationdistrict.childrensnational.org/wp-content/uploads/2025/01/Brats2024-Team-CNRI.jpg385685Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2025-01-10 17:12:152025-01-10 17:17:51AI for good: Children’s National wins global competitions for measuring brain tumors
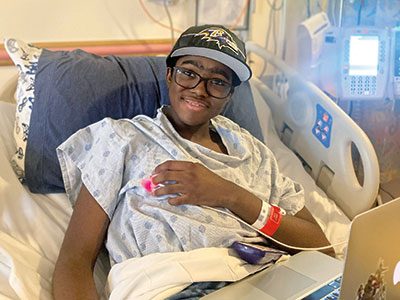
More than 100,000 Americans have sickle cell disease, an inherited blood disorder that can cause excruciating pain crises and shorter life expectancies.
Children’s National has one of the largest sickle cell programs in the United States. We are pioneering treatments and provide specialized care to about 1,500 patients each year. We participate in clinical trials to improve outcomes, shorten treatment time, reduce complications and minimize the need for opioids and chemotherapy.
Kendric receives care at Children’s National.
In recognition of our clinical and research excellence, Children’s National was one of a few U.S. pediatric hospitals selected to offer two promising new FDA-approved gene therapies.
Hematologist Robert Sheppard Nickel, M.D., leads a study to reduce toxicities in bone marrow transplants. “Years of development led to these curative therapies,” Dr. Nickel says. “I hope in the future we can safely cure more children with sickle cell disease.”
“The future looks promising to revolutionize the lives of our patients and make these therapies accessible worldwide,” says Andrew Campbell, M.D., director of our Comprehensive Sickle Cell Disease Program.
Kendric and Nasir find hope
In May 2024 at Children’s National, 12-year-old Kendric of Clinton, Maryland, became the world’s first patient with sickle cell disease to begin a commercially approved gene therapy that could dramatically reduce or even eliminate his pain. It involved extracting his bone marrow stem cells; genetically modifying them in a specialized lab to reduce the risk of sickling; and then, after chemotherapy, infusing them back into his bloodstream.
Expert, compassionate care empowered Kendric to understand the science behind his treatment and chart a path to recovery. “My care team taught me how to deal with my disease and everything that I need to know for the future,” he says. “They gave me hope that I could be cured.”
Nasir and his care team.
Nasir, age 20, spent his childhood waiting to find a match for a stem cell transplant to address his sickle cell disease. Finally, in 2023, at Children’s National, he found an answer in gene therapy to alter his own cells.
Due to painful episodes and the need for frequent blood transfusions, both Kendric and Nasir missed out on a lot of school, important moments with friends and simply being kids. Now, they can explore a world in which patients like themselves can overcome this disease and reclaim their health.
“I have all of this oxygen and energy that came out of nowhere,” Nasir says. “It’s really a new life. I feel reborn.”
“The network of doctors at Children’s National gave us reassurance and lots of hope,” says Kendric’s mom, Deborah. “They made us feel like family. We are in awe of how quickly things moved and how much compassion they have shown us.”
Andrew Dauber, M.D., M.M.Sc., chief of Endocrinology, with a patient.
Andrew Dauber, M.D., M.M.Sc., chief of Endocrinology, leads a program that brings together comprehensive resources for children with rare genetic growth disorders, including basic science, translational and clinical research.
“Discovery is important, and research gives us many answers,” he says. “But what we do with those answers is what really matters. There’s nothing better than seeing the impact our research is having on an individual patient. It is so gratifying to hear from parents about how their kids are making progress thanks to a protocol we developed.”
Mia thrives with the right approach
Middle schooler Mia loves to dance, practice gymnastics and hang out with friends. She was born in Croatia with hypochondroplasia, a genetic disorder that slows cell growth and causes short stature and limb shortening.
Mia practices her dance moves.
She struggled in kindergarten because she was so small. “Kids can be very mean if you’re different,” says Mia’s dad, Ivan. “For years, we took Mia to different specialists in Europe without getting the help she needed. I researched endocrinologists all over the world. They all pointed us to the United States.” This led Ivan to Children’s National and Dr. Dauber.
Dr. Dauber invited the family to the U.S. to participate in a clinical trial that was to start very soon. “I knew this was our only chance,” said Ivan, who rushed to bring his family to Washington, D.C., for the initial screening appointments. They later returned for more measurements, Mia’s first dose of medicine and a three-month supply to go. The family returns to Children’s National every six months.
In Mia’s first year, she grew more than 3 inches. Her arms also grew longer. “Dr. Dauber is probably the best doctor in the world,” Ivan says. “He is like a friend to Mia and has helped us make sure she has as normal a life as possible. This growth, including in her confidence, has been life-changing for her.” The family relocated to the U.S., and Mia will continue in the trial until she reaches puberty.
“Now, the difference between Mia and other kids is much less,” Ivan says. “She makes new friends more easily and is a happy, happy kid. As for me, I want to cry for how happy I am. Dr. Dauber and Children’s National made it all possible.”
https://innovationdistrict.childrensnational.org/wp-content/uploads/2024/12/dauber-and-patient-Feature.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2024-12-27 16:19:302024-12-27 16:20:43Innovative treatment for growth disorders
Dr. Seth Berger (right) and pediatric resident Dr. Kirkland Wilson.
Most patients with rare diseases still lack answers. Families may undergo years of searching in an often painful diagnostic odyssey.
Research by Seth Berger, M.D., Ph.D., a medical geneticist in our Center for Genetic Medicine Research and Rare Disease Institute, aims to harness technologies to shorten this journey and connect families with help sooner. Dr. Berger often publishes accounts of medical mysteries he has solved.
“It’s truly stunning what genetic sequencing can find. The outcomes can be life-changing. These cases with life-altering diagnoses don’t come along every day, but when they do, they make the hunt to find answers all the more worthwhile,” says Dr. Berger.
James finds a path to health
James, age 15, struggled a lot before a researcher at Children’s National found the needle in the haystack of his genome. Four years ago, he could not walk in a straight line down the sidewalk. Enjoying Halloween trick or treating in fall or a beach hike in summer? Out of the question. His gait had become increasingly unsteady.
Everything changed the day that Dr. Berger took a look at James’ exome — a subset of the genome that can reveal mutations — to help his family find answers. Dr. Berger used advanced biochemical testing, genomic sequencing and AI to sift through the patient’s data. He found the problem: dopa-responsive dystonia, a genetic condition seen in only one out of every 1 million children. In fact, James’ case was even rarer because he had an unusual recessive form.
James (left) rides mountain bikes with his brother, Nicholas, and mom, Shannon.
This discovery led to a cascade of positive events that transformed James’ life for the better. Thankfully, his condition has a known treatment. Laura Tochen, M.D., who directs the hospital’s Movement Disorders Program, started James on carbidopa-levodopa, a drug combination used to treat Parkinson’s disease and other neurological disorders. Within two hours, he showed improvement and his gait was almost normal.
Today, James leads an active life. On vacation last summer, he went rock climbing on real rocks for the first time. He loves mountain biking and running along the Maine coast. “He is very proud of what he can do now,” says his father, Jeff. “We are so thankful for the team that helped get him here.”
In 2024, Children’s National Hospital continued to make remarkable strides across diverse areas of pediatric medicine, from groundbreaking technological innovations to critical health advocacy. The following compilation showcases ten significant stories that demonstrate the breadth and depth of the hospital’s impact, as featured in major national news outlets including NBC Nightly News, CNN, The Washington Post, The New York Times, NPR, The Today Show, Healio, and POLITICO. Delve into our 2024 news highlights for more.
Charles Berul, M.D., and a patient family talk about the pill-sized pacemaker that saved the life of Abby, an infant born with deadly heart defects. (NBC Nightly News)
Sivabalaji Kaliamurthy, M.D., addiction psychiatrist and director of the Addictions Program, spoke to CNN about the impact of drug addiction on teen health and the lack of resources available to treat opioid use disorder. (CNN)
Susma Vaidya, M.D., M.P.H., associate medical director of the IDEAL Clinic, shared her concerns about childhood obesity treatment recommendations issued today by a leading panel of independent U.S. health experts. (The Washington Post)
Shideh Majidi, M.D., M.S.C.S., and Emily Frymark, clinical dietitian, spoke about how the food pharmacy, created in partnership with the Capital Area Food Bank, benefits patients with diabetes and other chronic conditions. (The Washington Post)
Kendric Cromer, a 12-year-old boy being treated at Children’s National Hospital, became the first person in the world with sickle cell disease to begin a commercially approved gene therapy that may cure the condition. “This is a big effort,” says David Jacobsohn, M.D., ScM, M.B.A. (The New York Times)
Mikael Petrosyan, M.D., associate chief of General and Thoracic Surgery, discusses the stress medical staff face when treating young victims of gun violence. (NPR)
Landon, an 11-year-old patient, rang the bell at Children’s National Hospital with family, friends, doctors and nurses cheering after finishing his final round of chemotherapy. (The Today Show)
Monika Goyal, M.D., M.S.C.E., pediatric emergency medicine specialist and co-director of the Center for Translational Research, emphasized the need for awareness in addressing period poverty in teenagers and young adults. (Healio)
Kolaleh Eskandanian, Ph.D., M.B.A., P.M.P., vice president and chief innovation officer, participates in a panel discussion covering AI data collection, associated risks, reliance and other topics related to artificial intelligence. (POLITICO)
Children’s National patient Kendric Cromer, 12, became one of the first children ever to be treated with a newly approved gene therapy that will free him from the sickle cell disease that has stolen his childhood. (The New York Times)
https://innovationdistrict.childrensnational.org/wp-content/uploads/2024/12/2024-News-Logo-Collage-feature.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2024-12-26 11:34:102024-12-26 11:49:26Children’s National in the News: 2024
2024 marked another groundbreaking year for Children’s National Hospital, showcasing remarkable advances across the spectrum of pediatric medicine, research and healthcare innovation. From pioneering surgical procedures to breakthrough artificial intelligence applications, the institution continued to push the boundaries of what’s possible in children’s healthcare. Read on for our list of the most popular articles we published on Innovation District in 2024.
A study led by researchers at Children’s National Hospital showed that babies born during the COVID-19 pandemic have differences in the size of certain structures in the brain, compared to infants born before the pandemic. The findings suggest that exposure to the coronavirus and being pregnant during the pandemic could play a role in shaping infant brain development. (3 min. read)
Children’s National Hospital was ranked as a top hospital in the nation by the U.S. News & World Report 2024-25 Best Children’s Hospitals annual rankings. This marks the eighth straight year Children’s National has made the Honor Roll list. The Honor Roll is a distinction awarded to only 10 children’s hospitals nationwide. (2 min. read)
In January 2023, a team of multidisciplinary doctors performed the first case in the world of using bilateral high intensity focused ultrasound (HIFU) pallidotomy on Jesus, a 22-year-old patient with dyskinetic cerebral palsy. The procedure is part of a clinical trial led by Chima Oluigbo, M.D., pediatric neurosurgeon at Children’s National Hospital. (3 min. read)
A novel ultrasound device developed by Bloom Standard received the Food and Drug Administration’s valued breakthrough device designation with the help of Children’s National Hospital. The device that enables autonomous, hands-free ultrasound scans to be performed anywhere, by any user. (2 min. read)
Understanding the effects of Lyme disease on the developing fetal brain is essential to ensure timely prenatal and postnatal treatments to protect the fetus and newborn. In response to this need, Children’s National Hospital is leading a pilot study to establish the groundwork needed for a larger study to determine the effect of in utero exposure to Lyme disease on pregnancy and early childhood neurodevelopmental outcomes. (3 min. read)
Five years ago, Cayden was born 6 weeks early weighing less than four pounds and at risk of dying from her critical congenital heart disease. Today, she’s a happy five-year-old. Early diagnosis of her hypoplastic right ventricle, double inlet left ventricle and critical coarctation of the aorta allowed for the team at Children’s National Hospital to create a careful plan for safe delivery and to offer an innovative hybrid HLHS surgical approach at the hospital within 24 hours after she was born. (1 min. read)
Children’s National Hospital appointed Wayne J. Franklin, M.D., F.A.C.C., as the new senior vice president (SVP) of the Children’s National Heart Center. In this role, Dr. Franklin oversees the full spectrum of heart care services including cardiac imaging and diagnostics, interventional cardiology, electrophysiology, cardiac anesthesia, cardiac surgery and cardiac intensive care. (2 min. read)
By pioneering artificial intelligence (AI) innovation programs at Children’s National Hospital, Marius George Linguraru, D.Phil., M.A., M.Sc., and the AI experts he leads are ensuring patients and families benefit from a coming wave of technological advances. The team is teaching AI to interpret complex data that could otherwise overwhelm clinicians. (4 min. read)
Painting a sobering picture, a research team led by Children’s National Hospital culled years of data demonstrating that maternal mental illness is an under-recognized contributor to the death of new mothers. They called for urgent action to address this public health crisis. (3 min. read)
Children’s National Hospital appointed Nathan Kuppermann, M.D., M.P.H., as its new executive vice president, chief academic officer and chair of Pediatrics. In this role, Dr. Kuppermann oversees research, education and innovation for the Children’s National Research Institute as well as academic and administrative leadership in the Department of Pediatrics at George Washington University School of Medicine & Health Services. (2 min. read)
Researchers from Children’s National Hospital presented findings from the first clinical trial of the medication vosoritide for children with hypochondroplasia – a rare genetic growth disorder. During the phase 2 trial, researchers found vosoritide increased the growth rate in children with hypochondroplasia, allowing them to grow on average an extra 1.8 cm per year. (2 min. read)
Since its establishment in July 2023, the Center for Prenatal, Neonatal & Maternal Health Research at Children’s National Hospital has gained recognition through high-impact scientific publications, featuring noteworthy studies exploring the early phases of human development. (3 min. read)
https://innovationdistrict.childrensnational.org/wp-content/uploads/2024/12/2024-with-lightbulb-CNRI.jpg385685Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2024-12-19 13:29:592024-12-19 13:33:52The best of 2024 from Innovation District
Nearly 200 biomedical leaders from Washington, D.C., Maryland, and Virginia gathered at the Children’s National Research & Innovation Campus for the 2nd annual Cell & Gene Therapy Symposium. The event showcased groundbreaking developments in rare disease treatments and underscored the importance of regional collaboration.
“By targeting diseases at the cellular level, we are on the cusp of breakthroughs in cell and gene therapy that will transform medicine,” said Catherine Bollard, M.D., M.B.Ch.B., director of the Center for Cancer and Immunology Research (CCIR) at Children’s National Hospital and a host of the symposium. “Progress will accelerate if we build partnerships beyond our own organizations.”
The big picture
Scientists and clinicians have worked for more than two decades to develop cell and gene therapies aimed at treating diseases on a cellular level. The past few years have been particularly promising as investment in science has led to advancements. Children’s National is at the forefront, as one of the first pediatric hospitals in the world to offer commercial gene therapies for sickle cell disease.
Many more treatments for rare diseases are in development at Children’s National and beyond. Leaders at CCIR are actively building collaborations with companies, academic institutions and enterprises across the mid-Atlantic region to accelerate these efforts.
During the symposium, Eugene Hwang, M.D., chief of Oncology at Children’s National, addressed the urgent need for more effective and less toxic treatments for pediatric brain tumors. He highlighted the potential of combining immunotherapies with innovations like low-intensity focused ultrasound, which can open the blood-brain barrier temporarily to improve drug delivery to tumors.
“With collaboration between the lab and clinic, alongside industry partners and even between hospitals, we can finally make strides I haven’t seen in my entire career,” Dr. Hwang said. “It’s an incredibly inspiring time for all of us.”
Why it matters
Experts from organizations as diverse as MaxCyte, ScaleReady, RoosterBio, PSC Biotech, Qiagen, FujiFilm and the Frederick County Office of Economic Development came together for the daylong conversation.
Michael Friedlander, Ph.D., executive director of the Fralin Biomedical Research Institute at Virginia Tech, emphasized the critical role of regional partnerships in fulfilling the potential of these emerging therapies. He pointed to the collaborative research between Children’s National and Virginia Tech on brain tumors, where bioengineers and cancer researchers are working side-by-side to create new treatments.
“We are now able to begin delivering these leading-edge therapies to patients,” Dr. Friedlander said. “For example, those who live in rural settings often have much less access to such frontline medical innovations. By collaborating with Children’s National and gaining access to urban pediatric populations, as well as patients in our more rural area, we can start to bring these therapies to a much broader audience.”
What’s next
Patrick Hanley, Ph.D., chief and director of the Cellular Therapy Program at Children’s National, observed that other regions in the U.S. are uniting to advance scientific discoveries with the backing of government, academia and industry. He hopes to see similar collaboration across the D.C., Maryland, and Virginia area, known as the DMV. Children’s National is leading an initiative called CHARM – the Capital Health and Mid-Atlantic Regenerative Medicine – to bring regional experts together for webinars, networking events and partnership opportunities.
“There’s significant interest in cell and gene therapy worldwide,” said Dr. Hanley, a symposium host. “I see an even greater interest in creating cell and gene therapy hubs. The time is right for our mid-Atlantic region, and I’m excited to see what unfolds in the next five years.”
https://innovationdistrict.childrensnational.org/wp-content/uploads/2024/11/CGT-Conference-CNRI.jpg385685Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2024-11-26 10:13:282024-11-26 10:15:14Regional powerhouse: Cell and Gene therapy leaders from mid-Atlantic forge connections
The research, published in Hormone Research in Paediatrics, examined whether pharmacokinetic (PK) factors or other blood biomarkers correlated with the response to treatment of vosoritide in children with hypochondroplasia.
Researchers from Children’s National Hospital presented new data from the world’s first clinical trial of vosoritide use for growth disorders at the European Society for Pediatric Endocrinology annual meeting. The research, published in Hormone Research in Paediatrics, examined whether pharmacokinetic (PK) factors or other blood biomarkers correlated with the response to treatment of vosoritide in children with hypochondroplasia. The study found that drug exposure — as measured by average PK area under the curve (AUC) — did not correlate with any growth outcome.
The big picture
Previously published results of the clinical trial of vosoritide in children with hypochondroplasia showed that vosoritide can help kids with hypochondroplasia grow better. For this study, the researchers analyzed PK parameters by non-compartmental methods. Pharmacodynamic markers were measured at day 1, the 6-month and 12-month visits. The PK analysis indicates that drug exposure was correlated to global C-type natriuretic peptide (CNP) activity as measured by urine cyclic guanosine monophosphate (cGMP) concentrations.
Moving the field forward
There are currently no approved therapies for hypochondroplasia. Children’s National is the first site in the world to conduct a trial of a targeted treatment for hypochondroplasia and this is the first study to report PKs of vosoritide in children with hypochondroplasia.
“The results of our study showed that the drug levels achieved are similar to what was achieved in patients with achondroplasia,” says Andrew Dauber, M.D., M.M.Sc., chief of Endocrinology at Children’s National and one of the study authors. “This suggests that the dose we used is a reasonable one, paving the way for a Phase 3 trial of vosoritide in children with hypochondroplasia.”
What’s next
Dr. Dauber emphasized the importance of these findings for the medical community, particularly for those dedicated to pediatric endocrinology. He noted that understanding the nuanced responses among different children is crucial for optimizing future treatments.
“This ongoing commitment to innovative research underscores the relentless pursuit of targeted therapies, bridging gaps and bringing hope to families and patients worldwide,” says Dr. Dauber.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2024/11/blood-test-feature.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2024-11-18 07:00:332024-11-15 14:17:40Pharmacokinetic and pharmacodynamic analysis of vosoritide in Phase 2 trial
Children’s National Hospital in Washington, D.C., was ranked as a top hospital in the nation by the U.S. News & World Report 2024-25 Best Children’s Hospitals annual rankings. This marks the eighth straight year Children’s National has made the Honor Roll list. The Honor Roll is a distinction awarded to only 10 children’s hospitals nationwide.
This year, U.S. News ended ordinal rankings on its Honor Roll. Instead of assigning a numerical rank from 1 to 10, all hospitals on the Honor Roll will be recognized as having attained the highest standards of care in the nation.
In addition, Children’s National tied for #1 pediatric hospital in the Mid-Atlantic region, which includes New York, New Jersey, Delaware, Pennsylvania, the District of Columbia, West Virginia and Virginia. It’s also best in the Mid-Atlantic in Neonatology.
For the fourteenth straight year, Children’s National ranked in 10 specialty services. New this year, U.S. News included behavioral health as a service line in the rankings. Since it’s the first year, there are no ordinal rankings for behavioral health, but the Children’s National program was named one of the top 50 programs in the country.
“In my first year here, I witnessed what makes Children’s National so special — our commitment to collaboration, empowering one another, and charting a bold path forward for pediatric care,” said Michelle Riley-Brown, MHA, FACHE, president and chief executive officer of Children’s National. “I’m proud U.S. News again recognized Children’s National as one of the top in the nation and the highest-ranked pediatric hospital in D.C., Maryland and Virginia. Together, we’ll continue to push the boundaries of care, research and innovation to make a difference for those who matter most — the kids.”
The annual rankings are the most comprehensive source of quality-related information on U.S. pediatric hospitals and recognizes the nation’s top 50 pediatric hospitals based on a scoring system developed by U.S. News.
“For nearly two decades, U.S. News has published Best Children’s Hospitals to empower the parents and caregivers of children with complex medical needs,” said Ben Harder, chief of health analysis and managing editor at U.S. News. “Children’s hospitals appearing on the U.S. News Honor Roll have a track record of delivering unparalleled specialized care.”
The bulk of the score for each specialty service is based on quality and outcomes data. The process includes a survey of relevant specialists across the country, who are asked to list hospitals they believe provide the best care for patients with the most complex conditions.
The Children’s National specialty services that U.S. News ranked in the top 10 nationally are:
https://innovationdistrict.childrensnational.org/wp-content/uploads/2024/10/US-News-Badges-2024-25-CNRI.jpg385685Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2024-10-08 01:00:002024-10-08 15:01:23Children’s National again ranked among the best in the nation by U.S. News & World Report
“It is a market failure that we are dealing with – a lack of incentives leading to a stagnation in innovation with respect to small markets, such as pediatrics. Children’s National Hospital and our partners in other children’s hospitals in the country play a critical role in making noise and sending a message that children should not be an afterthought.”
Hear more from Kolaleh Eskandanian, Ph.D., M.B.A., during her recent appearance at POLITICO. As vice president and chief innovation officer at Children’s National and Alliance for Pediatric Device Innovation principal investigator, Dr. Eskandanian shared her approach to engaging with the Food and Drug Administration (FDA) to advance artificial intelligence (AI) and machine learning technologies for pediatric healthcare. To date, she noted, the FDA has authorized 950 healthcare-related technologies enabled with AI and machine learning.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2024/10/Politico-CNRI.jpg385685Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2024-10-01 11:11:532025-03-10 13:30:17In the news: The future of patient care and access
His Highness Sheikh Mohamed bin Zayed Al Nahyan, President of the United Arab Emirates (right) visited Children’s National in September 2024.
Continuing a 30-year partnership that has yielded 82 U.S. patents and countless medical breakthroughs for kids and their families, the Government of the United Arab Emirates (UAE) has strengthened its transformational commitment to Children’s National Hospital with a new $35 million donation focused on prenatal, neonatal and maternal health.
The announcement of the new gift comes after a recent visit to the hospital by His Highness Sheikh Mohamed bin Zayed Al Nahyan, President of the United Arab Emirates (UAE), who met with Emirati families and patients receiving care at Children’s National Hospital.
The investment is the latest chapter of a longstanding philanthropic partnership between the UAE and Children’s National. Each year, more than 100 Emirati families travel to Children’s National for advanced pediatric care and life-saving treatments.
Researchers in the Center for Prenatal, Neonatal & Maternal Health Research are focused on the role of perinatal factors — including maternal stress, anxiety and depression — on the developing brain of the child. Studies also are revealing the impact of congenital anomalies such as heart disease and acquired conditions such as maternal infection with COVID-19 or Zika virus. New approaches to prenatal and postnatal care promise to optimize long-term outcomes of many hospitalized babies.
“Children in the Washington, D.C., area and across the world benefit greatly from the breakthroughs that have emerged from the incredible decades-long partnership between the UAE and Children’s National,” said Michelle Riley-Brown, President and CEO of Children’s National. “I am deeply grateful for the UAE’s most recent gift. The contribution will positively impact children and families and support the teams of researchers and specialists who dedicate their lives to developing innovative medical care.”
Key milestones
The UAE helped to establish the Sheikh Zayed Institute for Pediatric Surgical Innovation at Children’s National in 2009. Today, the Sheikh Zayed Institute (SZI) has grown into a world-class, self-sustaining research center receiving more than 80% of its funding from grants and outside sources.
This platform for invention is advancing autonomous, robotic surgery. The institute’s researchers believe pediatric surgical outcomes will improve if the precision and delicacy of a robot are incorporated into procedures such as gallbladder removal. SZI is also propelling the use of artificial intelligence to improve pediatric medicine and expand health equity. One example is a deep learning algorithm that uses hand-held ultrasounds to detect early signs of rheumatic heart disease, which kills nearly 400,000 people worldwide each year.
“The lives and health of countless children and families in the Washington area, in the UAE and around the world have been transformed by our partnership,” said Yousef Al Otaiba, the UAE Ambassador to the United States. “Our continued support promises even more breakthrough innovations in pediatric medicine.”
The UAE also supported the opening of the Children’s National Research & Innovation Campus through a 2019 commitment. The campus represents the first pediatric innovation hub of its kind, where scientists, inventors, caregivers, patients’ families and health authorities come together to advance pediatric health.
The Children’s National Rare Disease Institute and Center for Genetic Medicine Research are two of the teams housed at the campus. Together, they are pioneering care for children in the Washington region and abroad as an international referral site for rare disorders. Two examples of their research endeavors include: next-generation genomic testing to better understand how differences in genetic material can affect human health and identifying biochemical analytes.
The UAE opened a medical office in Washington, D.C., in 1991. Since then, thousands of Emirati patients have visited Children’s National for life-changing care for conditions such as congenital heart disease, neurological disorders and cancer. The hospital is currently treating 40 Emirati patients.
“Having our child treated at Children’s National means accessing specialized pediatric care from a renowned institution dedicated to children’s health,” said Hamad Alnuaimi, an Emirati father of a Children’s National patient. “It provides us with confidence and reassurance that our son is receiving the best possible medical attention from experts who understand and prioritize the unique needs of children. For the UAE to have a strong relationship with Children’s National signifies a valuable connection that enhances pediatric healthcare in our country. This partnership allows us to benefit from advanced treatments, medical innovations, and expertise that might otherwise be inaccessible. It represents a commitment to improving the health and well-being of children through international collaboration.”
https://innovationdistrict.childrensnational.org/wp-content/uploads/2024/09/UAE-visit-CNRI.jpg385685Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2024-09-26 09:00:002024-10-29 15:21:53New philanthropic support from the United Arab Emirates furthers research breakthroughs and care
Children’s National Hospital-affiliated participants will attend this year’s American Academy of Pediatrics National Conference and Exhibition. The meeting will take place in Orlando, Florida from September 27 to October 1. You will find a mini schedule of sessions below.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2024/09/AAP-2024-graphic-feature.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2024-09-19 18:24:202024-09-19 18:34:48Children’s National Hospital the 2024 American Academy of Pediatrics meeting
As researchers develop groundbreaking cellular therapies to treat pediatric solid tumors, physicians are preparing new ways to explain how these treatments work to patients and caregivers.
In a series of educational videos, scientists from Children’s National Hospital and institutions worldwide are offering tutorials on these novel treatments and how they target solid tumors at the cellular level.
“Let’s start by breaking down what a tumor-associated antigen-specific T (TAA-T) cell is,” Children’s National Research Technician Sammy Murphy says in one new video. “Our aim is to harness the power of T cells to identify and attack cancer cells.”
In less than six minutes, Murphy provides a short course on the details of these TAA-T cells and how her team combines their expertise in biology, medicine, bioinformatics, quality assurance and more to create the new therapies. “This collaborative team spirit has been a huge motivating factor and represents the best of what science can be,” she explains.
The big picture
Children’s National summer student Diana Kentell, a senior at Pratt Institute of Art studying digital art and 2D animation, created this video and a collection of others for the Cancer Grand Challenges (CGC), sponsored by the National Cancer Institute and Cancer Research UK. In 2022, the CGC awarded $25 million to Children’s National, the University College of London Cancer Institute and its partners on the NexTGen team to develop new therapies for pediatric solid tumors using CAR T cells.
The NexTGen team includes six patient advocates who have all been touched by pediatric tumors and support the scientists by providing the patient perspective on research and new treatments. These videos are a slice of the group’s efforts.
C. Russell Y. Cruz, M.D., Ph.D., a translational immunologist on the NexTGen team who oversaw Kentell’s video project, says bridging the gap between scientists and their patients who enroll in clinical trials will be essential to ensuring patients weigh the possibilities and the risks.
“Patient advocates help us understand our work from their perspective, making our science accessible to everyone,” Dr. Cruz said. “Engaging with such dedicated individuals often helps us refine our own ideas and provides invaluable insights. Most importantly, they remind us of our ultimate goal: to free future generations from the burden of pediatric cancer.”
Why we’re excited
In addition to the video on TAA-T cells, the team has assembled a collection of videos on killing assays, tumor slice assays and CAR T-cell manufacturing, which will help patients learn about the treatments when the clinical trials start. Sara Wakeling, who leads the NexTGen team’s patient advocates, said this toolkit will be a vital resource.
“Each of us came to this advocacy work because we were deeply affected by pediatric cancer. We aim to ensure that the child’s voice is central to the research and that the science is communicated in an informative and digestible way for patients’ families and the public,” Wakeling said. “With these videos, lay summaries of manuscripts and other explainers, we will have concrete information to share with families as soon as the new CAR T-cell therapies are ready for clinical trials.”
https://innovationdistrict.childrensnational.org/wp-content/uploads/2024/09/Cruz-video-still-CNRI.jpg385685Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2024-09-13 11:35:042024-09-13 11:36:45What’s a TAA-T? Advocates create videos to translate science for patients






























 2024 marked another groundbreaking year for Children’s National Hospital, showcasing remarkable advances across the spectrum of pediatric medicine, research and healthcare innovation. From pioneering surgical procedures to breakthrough artificial intelligence applications, the institution continued to push the boundaries of what’s possible in children’s healthcare. Read on for our list of the most popular articles we published on Innovation District in 2024.
2024 marked another groundbreaking year for Children’s National Hospital, showcasing remarkable advances across the spectrum of pediatric medicine, research and healthcare innovation. From pioneering surgical procedures to breakthrough artificial intelligence applications, the institution continued to push the boundaries of what’s possible in children’s healthcare. Read on for our list of the most popular articles we published on Innovation District in 2024.


 Children’s National Hospital in Washington, D.C., was ranked as a top hospital in the nation by the
Children’s National Hospital in Washington, D.C., was ranked as a top hospital in the nation by the 





