Voters select Children’s National innovation as runner-up in national competition

Facial recognition technology developed and tested by researchers with the Sheikh Zayed Institute for Pediatric Surgical Innovation and Rare Disease Institute at Children’s National was the runner-up in this year’s STAT Madness 2018 competition.
Facial recognition technology developed and tested by researchers with the Sheikh Zayed Institute for Pediatric Surgical Innovation and Rare Disease Institute at Children’s National was the runner up in this year’s STAT Madness 2018 competition. Garnering more than 33,000 overall votes in the bracket-style battle that highlights the best biomedical advances, the Children’s National entry survived five rounds and made it to the championship before falling short of East Carolina University’s overall vote count.
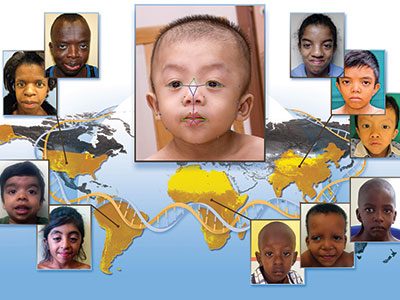
Children’s entry demonstrates the potential widespread utility of digital dysmorphology technology to diverse populations with genetic conditions. The tool enables doctors and clinicians to identify children with genetic conditions earlier by simply taking the child’s photo with a smartphone and having it entered into a global database for computer analyses.
The researchers partnered with the National Institutes of Health National Human Genome Research Institute and clinicians from 20 different countries to acquire pictures from local doctors for the study. Using the facial analysis technology, they compared groups of Caucasians, Africans, Asians and Latin Americans with Down syndrome, 22q11.2 deletion syndrome (also called DiGeorge syndrome) and Noonan syndrome to those without it. Based on more than 125 individual facial features, they were able to correctly identify patients with the condition from each ethnic group with more than a 93 percent accuracy rate. Missed diagnoses of genetic conditions can negatively impact quality of life and lead to premature death.
Children’s National also was among four “Editor’s Pick” finalists, entries that span a diverse range of scientific disciplines. Journalists at the digital publication STAT pored through published journal articles for 64 submissions in the single-elimination contest to honor a select group of entries that were the most creative, novel, and most likely to benefit the biomedical field and the general public.
Each year, 1 million children are born worldwide with a genetic condition that requires immediate attention. Because many of these children experience serious medical complications and go on to suffer from intellectual disability, it is critical that doctors accurately diagnose genetic syndromes as early as possible.
“For years, research groups have viewed facial recognition technology as a potent tool to aid genetic diagnosis. Our project is unique because it offers the expertise of a virtual geneticist to general health care providers located anywhere in the world,” says Marius George Linguraru, D.Phil., M.A., M.S., a Sheikh Zayed Institute for Pediatric Surgical Innovation principal investigator who invented the technology. “Right now, children born in under-resourced regions of the U.S. or the world can wait years to receive an accurate diagnosis due to the lack of specialized genetic expertise in that region.”
In addition to providing patient-specific benefits, Marshall Summar, M.D., director of Children’s Rare Disease Institute that partners in the facial recognition technology research, says the project offers a wider societal benefit.
“Right now, parents can endure a seemingly endless odyssey as they struggle to understand why their child is different from peers,” says Dr. Summar. “A timely genetic diagnosis can dispel that uncertainty and replace it with knowledge that can speed patient triage and deliver timely medical interventions.”