Carnitine may improve heart function in children receiving CRRT
A first-of-its-kind study demonstrated that IV carnitine supplementation is associated with improvement in myocardial strain and repletion of plasma total and free carnitine in children with AKI receiving CRRT.
Supplementation of a special nutrient could help improve heart function in children receiving continuous dialysis in critical care units. The nutrient carnitine plays an essential role in producing energy for use by heart and skeletal muscles. Critically ill children with acute kidney dysfunction often need a continuous dialysis therapy (also known as CRRT, continuous renal replacement therapy) to help remove toxins while kidneys are not working. An unintended consequence of this CRRT is removal of carnitine. Often these critically ill children are unable to eat by mouth and therefore can’t receive carnitine unless it is supplemented. Children’s National Hospital researchers have proven that intravenous carnitine supplementation is associated with repletion of the body’s carnitine supply and may cause improvement in heart function as shown by heart strain analysis (which detects subclinical cardiac dysfunction that may not be apparent by traditional echocardiography).
In a first-of-its-kind study, the Children’s National researchers, Asha Moudgil, M.D., Kristen Sgambat, M.D., and Sarah Clauss, M.D., investigated carnitine deficiency in children receiving CRRT. They demonstrated for the first time that these children become severely deficient in carnitine after being on CRRT for >1 week, and that carnitine supplementation is associated with carnitine repletion and improved heart function. This knowledge can help to guide clinical care, as carnitine can be easily added to the IV nutritional formulations that are typically given to these patients.
Although little was previously known about carnitine status in patients with acute kidney injury (AKI) receiving CRRT, iatrogenic carnitine deficiency related to chronic hemodialysis (HD) in patients with end stage renal disease is a well-known phenomenon. It was theorized that given the continuous removal of solutes by CRRT in combination with lack of dietary intake and impaired production of endogenous carnitine by the kidney in critically ill children with AKI, carnitine would be rapidly depleted.
The latest controlled pilot study (NCT01941823) of 48 children hypothesized that carnitine supplementation would improve left ventricular function in children receiving CRRT. Children ages 1-21 years with AKI requiring CRRT, who were admitted to the pediatric intensive care unit at Children’s National Hospital from 2015 to 2018 were eligible to prospectively enroll in the “CRRT Intervention group,” if they were total parenteral nutrition (TPN)-dependent and not receiving any enteral or IV carnitine prior to enrollment.
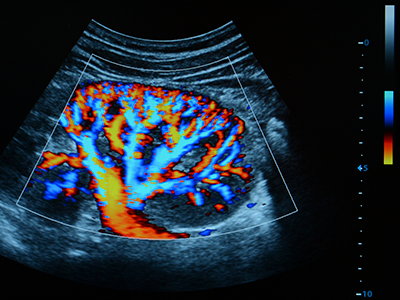
The researchers say that “An exciting collaborative effort between nephrology and cardiology made it possible to use a sophisticated technology known as speckle tracking imaging to study the effects of carnitine on heart in this population.” This technology can identify early changes in heart motion, also known as cardiac strain that may not be detected using standard heart imaging techniques.
This is the first study to demonstrate that IV carnitine supplementation is associated with improvement in myocardial strain and repletion of plasma total and free carnitine in children with AKI receiving CRRT. A cohort of pediatric chronic HD patients demonstrated similar benefits in a prior study conducted by Drs. Moudgil and Sgambat. Compared with chronic HD, carnitine is even more rapidly depleted by CRRT, with losses approximating 80% of intake. The effect of carnitine deficiency and supplementation on cardiovascular function in patients receiving CRRT had not been previously investigated.
The pilot study by Drs. Moudgil, Sgambat, and Clauss was single center and limited by small sample size. The small sample size may have limited the ability to detect significant differences in demographics and clinical characteristics and multivariable analyses could not be performed. However, given that it is a pilot study, the findings provide a solid launching point for future investigations to show how supplementation can be best utilized to optimize cardiac outcomes in children receiving CRRT.