Next-generation genomics testing holds key to undiagnosed rare disease
 Seth Berger, M.D., Ph.D., felt the pull to dig deeper when he started reading the chart. An 11-year-old boy had an abnormal gait and couldn’t even walk in a straight line down the sidewalk to go trick-or-treating. Yet workups with neurology, orthopedics and an exome analysis of the patient’s genetic code did not provide a diagnosis. He had been getting worse for roughly three years.
Seth Berger, M.D., Ph.D., felt the pull to dig deeper when he started reading the chart. An 11-year-old boy had an abnormal gait and couldn’t even walk in a straight line down the sidewalk to go trick-or-treating. Yet workups with neurology, orthopedics and an exome analysis of the patient’s genetic code did not provide a diagnosis. He had been getting worse for roughly three years.
With one of the largest clinical genetics departments in the country, Children’s National Hospital receives more than 10,000 visits a year from patients like this middle schooler. Often, they are children and caregivers who are searching for answers and follow-up support for diagnoses of genetic disorders, which impact so few people that only highly trained geneticists and genetic counselors can get to the root of the disorder.
“In genetics, we are finding layers of understanding. A negative clinical test is not always the final answer because the significance of variants can often be missed or misunderstood,” said Dr. Berger, a medical geneticist and principal investigator in the Center for Genetics Medicine Research at Children’s National. “It can take extensive research and a deep knowledge of the limits of certain tests to reach a diagnosis.”
The fine print
On page 4 of the patient’s genetics report, Dr. Berger found a reference to a pair of variants with no known clinical impact. Dr. Berger recognized that the genes referenced could affect proteins that drive potentially treatable neurological outcomes.
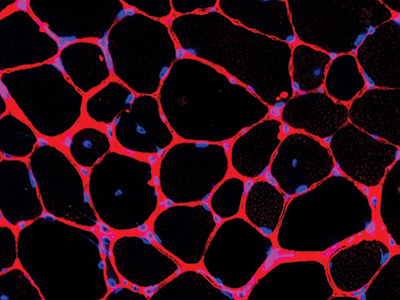
Dr. Berger ordered further testing, including biochemical testing of the patient’s blood and a phenylalanine loading challenge, a test that measures how the body metabolizes certain amino acids. With the results, he confirmed a recessive GCH1 deficiency in the patient was causing a condition called DOPA-responsive dystonia, a disorder that causes involuntary muscle contractions, tremors and uncontrolled movements. Laura Schiffman Tochen, M.D., director of the Movement Disorders Program at Children’s National, started the patient on levodopa-carbidopa — a drug combination used to treat Parkinson’s disease and other neurological disorders — and within two hours the boy showed improvement. His gait was almost normal.
Why we’re excited
Dr. Berger presents at conferences on this case and several other medical mysteries that he’s recently solved in his clinical practice and his role at the Pediatric Mendelian Genomics Research Center, a Children’s National program immersed in a federally funded research study to better understand how differences in genetic material can affect human health. As part of his work, he’s joined the GREGoR project (Genomic Research to Elucidate the Genetics of Rare Disease), which hopes to increase the number of genetic disorders where a cause can be identified. The elite genetics consortium includes nationally recognized research centers – the University of California at Irvine, Broad Institute, University of Washington, Baylor University, Stanford University, Invitae and Children’s National – which are working together to harness cutting-edge genomics sequencing capabilities. They hope to enroll thousands in their research, funded by the National Institutes of Health.
“It’s truly stunning what genetic sequencing can find. The outcomes can be life-changing,” said Dr. Berger. “These cases with life-altering diagnoses don’t come along every day, but when they do, they make the hunt to find answers all the more worthwhile.”


 The Children’s National Research Institute released its
The Children’s National Research Institute released its 
 Advanced MRI visualization techniques to follow blood flow in the hearts of cardiac patients. Gene therapy for pediatric patients with Duchenne muscular dystrophy. 3D-printed casts for treating clubfoot. These were among the most popular articles we published on Innovation District in 2023. Read on for our full list.
Advanced MRI visualization techniques to follow blood flow in the hearts of cardiac patients. Gene therapy for pediatric patients with Duchenne muscular dystrophy. 3D-printed casts for treating clubfoot. These were among the most popular articles we published on Innovation District in 2023. Read on for our full list.

 Unraveling how cells mend after injury serves as a key to unlocking potential therapies. Recent findings from the
Unraveling how cells mend after injury serves as a key to unlocking potential therapies. Recent findings from the