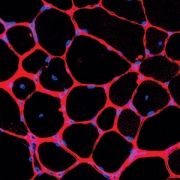
(A) Control population. (B) Population with Williams-Beuren syndrome. Average faces were generated for each demographic group after automatic face pose correction.
With an average accuracy of 88%, a deep learning technology offers rapid genetic screening that could accelerate the diagnosis of genetic syndromes, recommending further investigation or referral to a specialist in seconds, according to a study published in The Lancet Digital Health. Trained with data from 2,800 pediatric patients from 28 countries, the technology also considers the face variability related to sex, age, racial and ethnic background, according to the study led by Children’s National Hospital researchers.
“We built a software device to increase access to care and a machine learning technology to identify the disease patterns not immediately obvious to the human eye or intuition, and to help physicians non-specialized in genetics,” said Marius George Linguraru, D.Phil., M.A., M.Sc., principal investigator in the Sheikh Zayed Institute for Pediatric Surgical Innovation at Children’s National Hospital and senior author of the study. “This technological innovation can help children without access to specialized clinics, which are unavailable in most of the world. Ultimately, it can help reduce health inequality in under-resourced societies.”
This machine learning technology indicates the presence of a genetic syndrome from a facial photograph captured at the point-of-care, such as in pediatrician offices, maternity wards and general practitioner clinics.
“Unlike other technologies, the strength of this program is distinguishing ‘normal’ from ‘not-normal,’ which makes it an effective screening tool in the hands of community caregivers,” said Marshall L. Summar, M.D., director of the Rare Disease Institute at Children’s National. “This can substantially accelerate the time to diagnosis by providing a robust indicator for patients that need further workup. This first step is often the greatest barrier to moving towards a diagnosis. Once a patient is in the workup system, then the likelihood of diagnosis (by many means) is significantly increased.”
Every year, millions of children are born with genetic disorders — including Down syndrome, a condition in which a child is born with an extra copy of their 21st chromosome causing developmental delays and disabilities, Williams-Beuren syndrome, a rare multisystem condition caused by a submicroscopic deletion from a region of chromosome 7, and Noonan syndrome, a genetic disorder caused by a faulty gene that prevents normal development in various parts of the body.
Most children with genetic syndromes live in regions with limited resources and access to genetic services. The genetic screening may come with a hefty price tag. There are also insufficient specialists to help identify genetic syndromes early in life when preventive care can save lives, especially in areas of low income, limited resources and isolated communities.
“The presented technology can assist pediatricians, neonatologists and family physicians in the routine or remote evaluation of pediatric patients, especially in areas with limited access to specialized care,” said Porras et al. “Our technology may be a step forward for the democratization of health resources for genetic screening.”
The researchers trained the technology using 2,800 retrospective facial photographs of children, with or without a genetic syndrome, from 28 countries, such as Argentina, Australia, Brazil, China, France, Morocco, Nigeria, Paraguay, Thailand and the U.S. The deep learning architecture was designed to account for the normal variations in the face appearance among populations from diverse demographic groups.
“Facial appearance is influenced by the race and ethnicity of the patients. The large variety of conditions and the diversity of populations are impacting the early identification of these conditions due to the lack of data that can serve as a point of reference,” said Linguraru. “Racial and ethnic disparities still exist in genetic syndrome survival even in some of the most common and best-studied conditions.”
Like all machine learning tools, they are trained with the available dataset. The researchers expect that as more data from underrepresented groups becomes available, they will adapt the model to localize phenotypical variations within more specific demographic groups.
In addition to being an accessible tool that could be used in telehealth services to assess genetic risk, there are other potentials for this technology.
“I am also excited about the potential of the technology in newborn screening,” said Linguraru. “There are approximately 140 million newborns every year worldwide of which eight million are born with a serious birth defect of genetic or partially genetic origin, many of which are discovered late.”
Children’s National as well recently announced that it has entered into a licensing agreement with MGeneRx Inc. for its patented pediatric medical device technology. MGeneRx is a spinoff from BreakThrough BioAssets LLC, a life sciences technology operating company focused on accelerating and commercializing new innovations, such as this technology, with an emphasis on positive social impact.
“The social impact of this technology cannot be underestimated,” said Nasser Hassan, acting chief executive officer of MGeneRx Inc. “We are excited about this licensing agreement with Children’s National Hospital and the opportunity to enhance this technology and expand its application to populations where precision medicine and the earliest possible interventions are sorely needed in order to save and improve children’s lives.”




 Children’s National Hospital in Washington, D.C., was ranked #5 in the nation on the U.S. News & World Report 2023-24 Best Children’s Hospitals annual rankings. This marks the seventh straight year Children’s National has made the Honor Roll list. The Honor Roll is a distinction awarded to only 10 children’s hospitals nationwide.
Children’s National Hospital in Washington, D.C., was ranked #5 in the nation on the U.S. News & World Report 2023-24 Best Children’s Hospitals annual rankings. This marks the seventh straight year Children’s National has made the Honor Roll list. The Honor Roll is a distinction awarded to only 10 children’s hospitals nationwide.




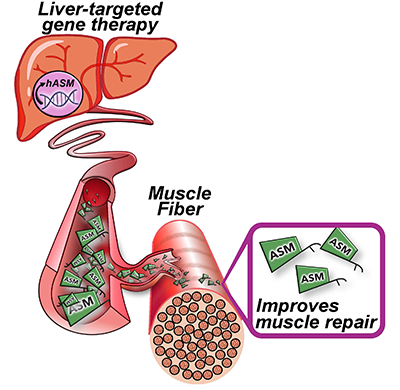
 A clinical trial testing a new drug to increase growth in children with short stature. The first ever high-intensity focused ultrasound procedure on a pediatric patient with neurofibromatosis. A low dose gene therapy vector that restores the ability of injured muscle fibers to repair. These were among the most popular articles we published on Innovation District in 2022. Read on for our full top 10 list.
A clinical trial testing a new drug to increase growth in children with short stature. The first ever high-intensity focused ultrasound procedure on a pediatric patient with neurofibromatosis. A low dose gene therapy vector that restores the ability of injured muscle fibers to repair. These were among the most popular articles we published on Innovation District in 2022. Read on for our full top 10 list.