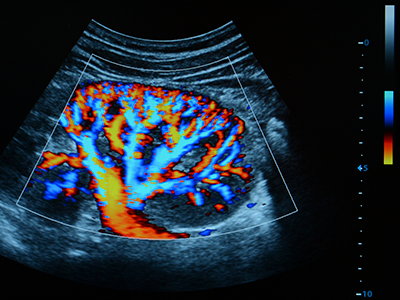
Researchers at Children’s National Hospital are using quantitative imaging and machine intelligence to enhance care for children with a common kidney disease, and their initial results are very promising. Their technique provides an accurate way to predict earlier which children with hydronephrosis will need surgical intervention, simplifying and enhancing their care.
We live in a time of great uncertainty yet great promise, particularly when it comes to harnessing technology to improve lives. Researchers at Children’s National Hospital are using quantitative imaging and machine intelligence to enhance care for children with a common kidney disease, and their initial results are very promising. Their technique provides an accurate way to predict earlier which children with hydronephrosis will need surgical intervention, simplifying and enhancing their care.
Hydronephrosis means “water in the kidney” and is a condition in which a kidney doesn’t empty normally. One of the most frequently detected abnormalities on prenatal ultrasound, hydronephrosis affects up to 4.5% of all pregnancies and is often discovered prenatally or just after birth.
Although hydronephrosis in children sometimes resolves by itself, identifying which kidneys are obstructed and more likely to need intervention isn’t particularly easy. But it is critical. “Children with severe hydronephrosis over long periods of time can start losing kidney function to the point of losing a kidney,” says Marius George Linguraru, DPhil, MA, MSc, principal investigator of the project; director of Precision Medical Imaging Group at the Sheikh Zayed Institute for Pediatric Surgical Innovation; and professor of radiology, pediatrics and biomedical engineering at George Washington University.
Children with hydronephrosis face three levels of examination and intervention: ultrasound, nuclear imaging testing called diuresis renogram and surgery for the critical cases. “What we want to do with this project is stratify kids as early as possible,” Dr. Linguraru says. “The earlier we can predict, the better we can plan the clinical care for these kids.”
Ultrasound is used to see whether there is a blockage and try to determine hydronephrosis severity. “Ultrasound is non-invasive, non-radiating, and does not expose the child to any risk prenatally or postnatally,” Dr. Linguraru says. Ultrasound evaluations require a trained radiologist, but there’s a lot of variability. Radiologists have a grading system based on the ultrasound appearance of the kidney to determine whether the hydronephrosis is mild, moderate or severe, but studies show this isn’t predictive of longer term outcomes.
Children whose ultrasounds show concern will be referred to diuresis renogram. Costly, complex, invasive and irradiating, it tests how well the kidney empties. Although appropriate for good clinical indications, doctors try to minimize its use. “Management of hydronephrosis is complex,” Dr. Linguraru says. “We want to use ultrasound as much as possible and much less diuresis renogram.”
For those patients whose kidney is obstructed and eventually need surgical intervention, the sooner that decision can be made the better. “The more you wait for a kidney that is severely obstructed, the more function may be lost. If intervention is required, it’s preferable to do it early,” Dr. Linguraru says. Of course for the child whose hydronephrosis will likely resolve itself, intervention is not the best option.

“With our technique we are measuring physiological and anatomical changes in the ultrasound image of the kidney,” says Marius George Linguraru, DPhil, MA, MSc. “The human eye may find it difficult to put all this together, but the machine can do it. We use quantitative imaging to do deep phenotyping of the kidney and machine learning to interpret the data.”
Dr. Linguraru and the multidisciplinary team at Children’s National Hospital, including radiology and urology clinicians, are putting the power of computers to work interpreting subtleties in the ultrasound data that humans just can’t see. In their pilot study they found that 60% of the nuclear imaging tests could have been safely avoided without missing any of the critical cases of hydronephrosis. “With our technique we are measuring physiological and anatomical changes in the ultrasound image of the kidney,” Dr. Linguraru says. “The human eye may find it difficult to put all this together, but the machine can do it. We use quantitative imaging to do deep phenotyping of the kidney and machine learning to interpret the data.”
Results of the initial study indicate that kids who have a mild condition can be safely discharged earlier and the model can predict all those kids with obstructions and accelerate their diagnosis by sending them earlier to get further investigation. Dr. Linguraru says. “There are only benefits: some kids will get earlier diagnosis, some earlier discharges.”
The team also has a way to improve the interpretation of diuresis renograms. “We analyze the dynamics of the kidney’s drainage curve in quantifiable way. Using machine learning to interpret those results, we showed we can potentially discharge some kids earlier and accelerate intervention for the most severe cases instead of waiting and repeating the invasive tests,” he says. The framework has 93% accuracy, including 91% sensitivity and 96% specificity, to predict surgical cases, a significant improvement over clinical metrics’ accuracy.
The next step is a study connecting all the protocols. “Right now we have a study on ultrasound, a study on nuclear imaging, but we need to connect them so a child with hydronephrosis immediately benefits,” says Dr. Linguraru. Future work will focus on streamlining and accelerating diagnosis and intervention for kids who need it, both in prospective studies and hopefully clinically as well.
Hydronephrosis is an area in which machine learning can be applied to pediatric health in meaningful ways because of the sheer volume of cases.
“Machine learning algorithms work best when they are trained well on a lot of data,” Dr. Linguraru says. “Often in pediatric conditions, data are sparse because conditions are rare. Hydronephrosis is one of those areas that can really benefit from this new technological development because there is a big volume of patients. We are collecting more data, and we’re becoming smarter with these kinds of algorithms.”
Learn more about the Precision Medical Imaging Laboratory and its work to enhance clinical information in medical images to improve children’s health.



 Thousands of medical professionals and researchers from around the world gathered in San Diego this October for
Thousands of medical professionals and researchers from around the world gathered in San Diego this October for 











 Advanced MRI visualization techniques to follow blood flow in the hearts of cardiac patients. Gene therapy for pediatric patients with Duchenne muscular dystrophy. 3D-printed casts for treating clubfoot. These were among the most popular articles we published on Innovation District in 2023. Read on for our full list.
Advanced MRI visualization techniques to follow blood flow in the hearts of cardiac patients. Gene therapy for pediatric patients with Duchenne muscular dystrophy. 3D-printed casts for treating clubfoot. These were among the most popular articles we published on Innovation District in 2023. Read on for our full list.




 Children’s National Hospital is joining the International Pediatric Nephrology Association (IPNA) to bring care to children with kidney disease in Jamaica. With early screenings, diagnosis and optimal treatments, this collaboration will help decrease the morbidity and mortality associated with renal disease.
Children’s National Hospital is joining the International Pediatric Nephrology Association (IPNA) to bring care to children with kidney disease in Jamaica. With early screenings, diagnosis and optimal treatments, this collaboration will help decrease the morbidity and mortality associated with renal disease.