
Behind the Nociometer: Q&A with Julia Finkel, MD
Julia Finkel, MD, is a pediatric anesthesiologist and the director of Pain Medicine Research at the Sheikh Zayed Institute for Pediatric Surgical Innovation.
What if doctors could measure pain as precisely as they measure blood pressure? That’s the vision driving Julia Finkel, MD, a pediatric anesthesiologist and director of Pain Medicine Research at the Sheikh Zayed Institute for Pediatric Surgical Innovation. Recently featured in The Washington Post, Dr. Finkel is pioneering the Nociometer, a new device that could transform how we understand and treat pain.
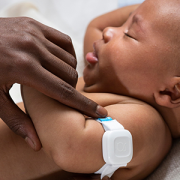
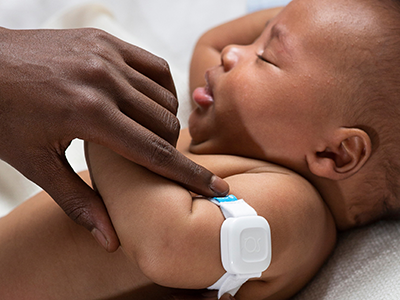
This new innovation uses a painless electrical current on a finger or toe to gently stimulate the body’s three main sensory nerve fibers. It then measures pupil response — linked to the brain’s pain centers — to identify the type and intensity of pain.
Now heading into clinical trials, the Nociometer shows how federal funding fuels innovation for patients. Here, Dr. Finkel shares the inspiration behind her work and the vital role of federal support in pediatric research.
Q: What started you on the path to developing this device?
A: My father is a theoretical physicist. He often talked in lay terms about everything he was thinking. When I was a child, we’d go for walks at night and look at the stars, and he’d explain the universe. My mother was an artist, but she struggled with constant pain from rheumatoid arthritis. There weren’t very good therapeutics at the time. Pain impacted her quality of life.
As an undergraduate, I was a chemistry major and interested in the mechanisms of how things worked. I planned to be a rheumatologist, but switching to anesthesia was transformative.
Q: What did you love about anesthesiology?
A: I love how it profoundly impacts patients. You induce unconsciousness, you induce wakefulness and you eliminate pain. When pain stops for a child, it also lessens the suffering of the parents. Our research is all about gaining a deeper understanding of the mechanisms of pain.
The Nociometer helps identify the mechanism behind the pain symptom so we can treat it appropriately. Research shows us that pain has a recognizable signature in the body. Because we can recognize it, we can treat it better. It’s like having an X-ray to look at before treating a broken leg.
Q: What is it like to work with children experiencing chronic pain? Why is measuring pain this way more effective than the typical scale?
A: Families become desperate. Often, they have seen many doctors before coming to a specialist. But pain is a single word for many different disease states. Not everyone has the right expertise. We often see children with chronic abdominal pain. Perhaps they struggle in school or can’t go at all. They don’t eat well, they have trouble engaging in regular activities. The Nociometer helps us determine the cause of the pain to better treat the problem. The device also gives us a way to validate pain while the pain scale tells us perception. The Nociometer provides objective data, very similar to a blood pressure reading.
Q: How has federal funding, and support from Children’s National, shaped your work?
A: Federal funding has been absolutely critical. Initially, we had grants from multiple agencies within the National Institutes of Health (NIH). This shows how ubiquitous and problematic pain is across the board. It’s the underpinning of many disease states and an upstream driver of the opioid epidemic.
We have grants from the Small Business Innovation Research program within the National Cancer Institute. We have funding from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and an award from the Advanced Research Projects Agency for Health. This federal funding comes with scientific rigor and the ecosystem at Children’s National has allowed me to accomplish so much.
Q: What makes Children’s National the right place for this kind of innovation?
A: Children’s National has given me the space to think creatively and pursue unconventional ideas. In 2011, I became a founding member of the Sheikh Zayed Institute for Pediatric Surgical Innovation. With early institutional support, I was able to explore questions that led to the discovery of a physiological biomarker for pain — the basis of the Nociometer.
“Creative” isn’t a word you often hear in medicine, but here, it’s part of the process. I was able to spin out a company, secure funding and build a prototype — steps that wouldn’t have been possible elsewhere. The culture at Children’s National embraces discovery and fuels real impact.
Julia Finkel, MD, is a pediatric anesthesiologist and Director of Pain Medicine Research and Development, The Sheikh Zayed Institute for Pediatric Surgical Innovation and Professor of Anesthesiology, Pediatrics and Critical Care Medicine at The George Washington University.












 Adapted from
Adapted from 

 Many service providers struggle to keep pace with advances in autism-specific knowledge and tend to refer children to autism specialty clinics when the diagnosis of autism spectrum disorder (ASD) is in question. Unfortunately, it is in these settings where children most often wait for months or, worse, experience barriers to accessing any care at all. This has resulted in an access crisis for children and families with ASD concerns contributing to delays in diagnosis and treatment, particularly for children of color and for under-resourced families. Service disruptions and challenges related to the COVID-19 pandemic have only added to delays. As the need for autism-related services continues to grow, innovative models must be used to enhance competence among frontline medical, behavioral health and community-based providers who currently serve these children and families on a regular basis.
Many service providers struggle to keep pace with advances in autism-specific knowledge and tend to refer children to autism specialty clinics when the diagnosis of autism spectrum disorder (ASD) is in question. Unfortunately, it is in these settings where children most often wait for months or, worse, experience barriers to accessing any care at all. This has resulted in an access crisis for children and families with ASD concerns contributing to delays in diagnosis and treatment, particularly for children of color and for under-resourced families. Service disruptions and challenges related to the COVID-19 pandemic have only added to delays. As the need for autism-related services continues to grow, innovative models must be used to enhance competence among frontline medical, behavioral health and community-based providers who currently serve these children and families on a regular basis.
 The choice between stereoelectroencephalography (SEEG) and subdural evaluation is not mutually exclusive, according to a new
The choice between stereoelectroencephalography (SEEG) and subdural evaluation is not mutually exclusive, according to a new 