
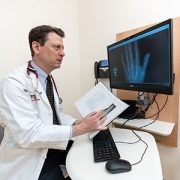
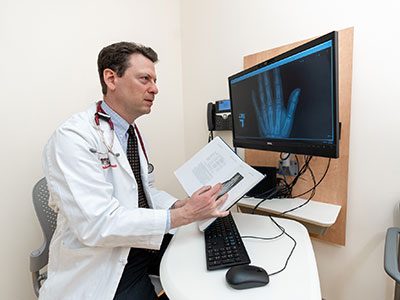
Children with hypochondroplasia were previously left with no options for effective long-term treatment, but Andrew Dauber, MD, MMSc, and his team continue to see promising results for treating children with hypochondroplasia using vosoritide.
For many children with short stature and other rare genetic growth disorders, there have been no next steps after usual treatment options prove ineffective. Researchers at Children’s National Hospital are digging deeper to find the root genetic causes of short stature disorders and creating novel, nuanced treatment options that have the opportunity to change how the field approaches these cases. From the creation of the growth specialty clinic to creating a study for one patient, the endocrinology team at Children’s National is focused on treating children with the uniqueness that their growth disorders require.
Many short stature disorders are caused by genetic variations that slow the growth of cells in the growth plate.
Children with hypochondroplasia were previously left with no options for effective long-term treatment, but Andrew Dauber, MD, MMSc, chief of Endocrinology at Children’s National, and his team continue to see promising results for treating children with hypochondroplasia using vosoritide. This drug had previously been approved for treatment of children with achondroplasia, the most common form of dwarfism. Treatment of hypochondroplasia has shown similar and, often, even better results. Study participants have managed the treatment well and have overall been very satisfied with the results.
Children’s National is the only site in the world offering this treatment to patients. The study includes over 50 children from two distinct subsets of patients — those with hypochondroplasia and those with other genetic short stature disorders such as RASopathy conditions, most common of which is Noonan syndrome, and children with mutations in the aggrecan and NPR2 genes. The study has found a significant increase in growth rates for children with hypochondroplasia who underwent treatment with vosoritide for one year.
“These are the first patients in the world to ever receive this medication for their conditions,” said Dr. Dauber. “The results are very promising and may change the way we practice medicine. Patients have come from all over the world to participate in the study.”
The preliminary data is even more promising for children in the study with other genetic conditions, which include defects that are more directly related to C-natriuretic peptide (CNP), which vosoritide targets directly. This is the first medication that directly targets the pathway in chondrocytes (cells in the growth plate that make the bones grow longer) affected by these specific mutations. Those patients are still undergoing their first full year of treatment and results of that section of the study are expected to be released next fall.
“We’re really starting to see this therapeutic landscape open up and develop for patients with this rare condition, for which right now there is no approved therapy, so really exciting times in this space,” says Dr. Dauber.
Dr. Dauber was a part of a study that was the first-of-its-kind to provide genetic testing for children with short stature and their families, finding ACAN gene mutations in multiple family members, and providing hormone therapy to the children impacted. The ability to diagnose this type of gene mutation allows families to be proactive with treatments — both as their child is growing and how other family members can reduce the risk of further complications down the road.
“We’re at the tip of the iceberg with research that explores this gene mutation,” says Dr. Dauber.
This type of unique and cutting-edge research isn’t new to the endocrinology team at Children’s National, who is focused on creating unique interventions to find answers for patients and families. When a patient with short stature was found to have a unique mutation in his growth hormone receptor, Dr. Dauber and his team created a single patient trial with a precision medicine approach to overcome the patient’s growth hormone resistance. This isn’t the first time Dr. Dauber led a single patient study. Even when study populations are small, unlocking genetic answers and treatment options for even one patient is at the core of work being done at Children’s National. Dr. Dauber emphasized the importance of these findings for the medical community, particularly for those dedicated to pediatric endocrinology. He noted that understanding the nuanced responses among different children is crucial for optimizing future treatments.
Even with improved height and growth outcomes, there is still more to uncover. In addition to the world’s first clinical trial using vosoritide in children for hypochondroplasia, Children’s National researchers are studying the quality of life for children with the disorder and how it may be affected by treatment, aiming to provide this full scope of care for children with these rare conditions. Dr. Dauber and his team continue to study connections between genetic biomarkers and response rates to clinical therapies, with hopes of discovering how these targeted approaches to treatment can be most effective. The work done by the team at Children’s National has shown results to warrant phase three of the trial.
“This ongoing commitment to innovative research underscores the relentless pursuit of targeted therapies, bridging gaps and bringing hope to families and patients worldwide,” says Dr. Dauber.


Andrew Dauber, M.D., M.M.Sc., chief of Endocrinology, with a patient.
Andrew Dauber, M.D., M.M.Sc., chief of Endocrinology, leads a program that brings together comprehensive resources for children with rare genetic growth disorders, including basic science, translational and clinical research.
“Discovery is important, and research gives us many answers,” he says. “But what we do with those answers is what really matters. There’s nothing better than seeing the impact our research is having on an individual patient. It is so gratifying to hear from parents about how their kids are making progress thanks to a protocol we developed.”
Middle schooler Mia loves to dance, practice gymnastics and hang out with friends. She was born in Croatia with hypochondroplasia, a genetic disorder that slows cell growth and causes short stature and limb shortening.

Mia practices her dance moves.
She struggled in kindergarten because she was so small. “Kids can be very mean if you’re different,” says Mia’s dad, Ivan. “For years, we took Mia to different specialists in Europe without getting the help she needed. I researched endocrinologists all over the world. They all pointed us to the United States.” This led Ivan to Children’s National and Dr. Dauber.
Dr. Dauber invited the family to the U.S. to participate in a clinical trial that was to start very soon. “I knew this was our only chance,” said Ivan, who rushed to bring his family to Washington, D.C., for the initial screening appointments. They later returned for more measurements, Mia’s first dose of medicine and a three-month supply to go. The family returns to Children’s National every six months.
In Mia’s first year, she grew more than 3 inches. Her arms also grew longer. “Dr. Dauber is probably the best doctor in the world,” Ivan says. “He is like a friend to Mia and has helped us make sure she has as normal a life as possible. This growth, including in her confidence, has been life-changing for her.” The family relocated to the U.S., and Mia will continue in the trial until she reaches puberty.
“Now, the difference between Mia and other kids is much less,” Ivan says. “She makes new friends more easily and is a happy, happy kid. As for me, I want to cry for how happy I am. Dr. Dauber and Children’s National made it all possible.”

The research, published in Hormone Research in Paediatrics, examined whether pharmacokinetic (PK) factors or other blood biomarkers correlated with the response to treatment of vosoritide in children with hypochondroplasia.
Researchers from Children’s National Hospital presented new data from the world’s first clinical trial of vosoritide use for growth disorders at the European Society for Pediatric Endocrinology annual meeting. The research, published in Hormone Research in Paediatrics, examined whether pharmacokinetic (PK) factors or other blood biomarkers correlated with the response to treatment of vosoritide in children with hypochondroplasia. The study found that drug exposure — as measured by average PK area under the curve (AUC) — did not correlate with any growth outcome.
Previously published results of the clinical trial of vosoritide in children with hypochondroplasia showed that vosoritide can help kids with hypochondroplasia grow better. For this study, the researchers analyzed PK parameters by non-compartmental methods. Pharmacodynamic markers were measured at day 1, the 6-month and 12-month visits. The PK analysis indicates that drug exposure was correlated to global C-type natriuretic peptide (CNP) activity as measured by urine cyclic guanosine monophosphate (cGMP) concentrations.
There are currently no approved therapies for hypochondroplasia. Children’s National is the first site in the world to conduct a trial of a targeted treatment for hypochondroplasia and this is the first study to report PKs of vosoritide in children with hypochondroplasia.
“The results of our study showed that the drug levels achieved are similar to what was achieved in patients with achondroplasia,” says Andrew Dauber, M.D., M.M.Sc., chief of Endocrinology at Children’s National and one of the study authors. “This suggests that the dose we used is a reasonable one, paving the way for a Phase 3 trial of vosoritide in children with hypochondroplasia.”
Dr. Dauber emphasized the importance of these findings for the medical community, particularly for those dedicated to pediatric endocrinology. He noted that understanding the nuanced responses among different children is crucial for optimizing future treatments.
“This ongoing commitment to innovative research underscores the relentless pursuit of targeted therapies, bridging gaps and bringing hope to families and patients worldwide,” says Dr. Dauber.
Read the study in Hormone Research in Paediatrics: Phase 2 Trial of Vosoritide Use in patients with Hypochondroplasia: Pharmacokinetic/ Pharmacodynamic analysis from 12 Month Data
A patient with short stature and growth hormone (GH) resistance experienced a growth rate increase of 3.4 cm/year after 12 months of high-dose GH therapy.
In a first-of-its-kind single-patient clinical trial, a patient with short stature and growth hormone (GH) resistance experienced a growth rate increase of 3.4 cm/year after 12 months of high-dose GH therapy as part of a precision medicine approach. The study, led by Andrew Dauber, M.D., M.M.Sc., chief of Endocrinology at Children’s National Hospital, was published in The Journal of Clinical Endocrinology & Metabolism.
“Many patients with short stature do not get a definitive genetic diagnosis, and even if they do, it takes substantial effort to design a targeted therapeutic trial,” says Dr. Dauber.
This is the first trial of extremely high-dose growth hormone in a single patient with a unique form of growth hormone resistance.
During an endocrine evaluation for short stature, which included GH stimulation testing demonstrating growth hormone resistance, a next-generation sequencing panel revealed that the patient had an inherited heterozygous frameshift variant in the growth hormone receptor gene (GHR).
The researchers found that the patient’s growth hormone binding protein (GHBP) levels were elevated because of this variant, limiting circulating GH’s ability to bind to the GH receptor. The researchers believed that, with extremely high doses of GH, the mechanism of resistance would be overcome and normal GH signaling would be restored.
“We found that we were able to overcome the patient’s growth hormone resistance, which resulted in substantially improved growth,” says Dr. Dauber. “Our understanding of the underlying pathophysiology of his growth disorder led to our ability to design a precision medicine trial just for this single patient.”
Genetic testing and translational biology can provide insights into the specific underlying mechanism of rare growth disorders, allowing for precision medicine approaches.


Trial participant Mia Maric is measured by Dr. Andrew Dauber.
Researchers from Children’s National Hospital presented findings from the first clinical trial of the medication vosoritide for children with hypochondroplasia – a rare genetic growth disorder. The results were presented at the 2024 American College of Medical Genetics and Genomics (ACMG) Annual Clinical Genetics Meeting.
Recently approved to increase linear growth and open growth plates in children with achondroplasia, vosoritide is a C-type natriuretic peptide analog that binds its receptor on chondrocytes, leading to increased chondrocyte proliferation and differentiation by inhibiting the ERK1/2-MAPK pathway.
“Vosoritide directly targets the pathway in the growth plate that is affected by the genetic mutation causing hypochondroplasia,” said Andrew Dauber M.D., M.M.Sc., chief of Endocrinology at Children’s National and lead author of the study.
During the phase 2 trial, researchers found vosoritide increased the growth rate in children with hypochondroplasia, allowing them to grow on average an extra 1.8 cm per year.
Ivan Maric’s 11-year-old daughter, Mia, has been participating in the trial for the last 18 months. In 2022, they moved from Croatia to be part of the study.
“This has been life-changing for Mia,” Maric said. “Soon after receiving the initial doses, we immediately noticed growth. Now, she can independently manage everyday tasks such as washing her hair and reaching the sink to wash her hands.”
Vosoritide treatment may work as a precision therapy to improve growth in multiple genetic conditions that interact with the ERK1/2-MAPK pathway.
“This study provides a proof of principle that this medicine will work for these children and supports further research in this area,” said Dr. Dauber. “I was excited to see how well tolerated the medication was and how some patients had excellent responses.”
This clinical trial funded by BioMarin is the first-of-its-kind to treat children with genetic short stature who do not have achondroplasia. Other growth-related conditions included in this phase 2 trial were Noonan syndrome, NPR2 mutations and Aggrecan mutations.
Additional authors from Children’s National: Anqing Zhang, Ph.D., Roopa Kanakatti Shankar, M.D., Kimberly Boucher, R.N., Tara McCarthy, B.A., Niusha Shafaei, B.A., Raheem Seaforth, B.A., Meryll Grace Castro, M.S., and Niti Dham, M.D.


Children with hypochondroplasia have low parent-reported quality of life (QOL) scores, according to findings from researchers at Children’s National Hospital.
Children with hypochondroplasia have low parent-reported quality of life (QOL) scores, according to findings from researchers at Children’s National Hospital. The data, presented as part of a poster presentation at the Pediatric Endocrine Society (PES) annual meeting, also found older age and shorter height may further exacerbate effects on QOL.
Hypochondroplasia is a skeletal dysplasia that has an estimated prevalence of 1 in 15,000-40,000 births and is characterized by short stature and disproportionately short arms, legs, hands and feet.
Participants of the study were 13 prepubertal boys ages 3-11 and 13 prepubertal girls ages 3-10, all with height z-scores < -2.25 SD and genetically proven hypochondroplasia.
Effective medications for growth in patients with hypochondroplasia are limited. Children’s National is participating in an ongoing study of a new drug, vosoritide, used to treat children in this population and will compare pre- and post-intervention QOL scores.
Understanding QOL of children with hypochondroplasia will help clinicians provide better care and support.
“Knowing how a medication affects QOL will help guide counseling about use of this medication,” says Nicole Rangos, M.D., pediatric resident at Children’s National and lead author of the study.
Dr. Rangos received the Human Growth Foundation Award for best growth-related abstract at the PES annual meeting in May 2023. The award was established in 2007 to encourage fellows training in pediatric endocrinology to pursue clinical and bench investigations that may lead to a better understanding of human growth.

 A clinical trial testing a new drug to increase growth in children with short stature. The first ever high-intensity focused ultrasound procedure on a pediatric patient with neurofibromatosis. A low dose gene therapy vector that restores the ability of injured muscle fibers to repair. These were among the most popular articles we published on Innovation District in 2022. Read on for our full top 10 list.
A clinical trial testing a new drug to increase growth in children with short stature. The first ever high-intensity focused ultrasound procedure on a pediatric patient with neurofibromatosis. A low dose gene therapy vector that restores the ability of injured muscle fibers to repair. These were among the most popular articles we published on Innovation District in 2022. Read on for our full top 10 list.
Preliminary results from a phase II clinical trial at Children’s National Hospital showed that a new drug, vosoritide, can increase growth in children with certain growth disorders. This was the first clinical trial in the world testing vosoritide in children with certain genetic causes of short stature.
(2 min. read)
Children’s National Hospital successfully performed the first ever high-intensity focused ultrasound (HIFU) non-invasive procedure on a pediatric patient with neurofibromatosis. This was the youngest patient to undergo HIFU treatment in the world.
(3 min. read)
Using a single injection of a low dose gene therapy vector, researchers at Children’s National restored the ability of injured muscle fibers to repair in a way that reduced muscle degeneration and enhanced the functioning of the diseased muscle.
(3 min. read)
A world-class team of researchers co-led by Catherine Bollard, M.D., M.B.Ch.B., director of the Center for Cancer and Immunology Research at Children’s National, was selected to receive a $25m Cancer Grand Challenges award to tackle solid tumors in children.
(4 min. read)
Children’s National opened a new telehealth command center that uses cutting-edge technology to keep continuous watch over children with critical heart disease. The center offers improved collaborative communication to better help predict and prevent major events, like cardiac arrest.
(2 min. read)
Children’s National named Monika Goyal, M.D., M.S.C.E., associate chief of Emergency Medicine, as the first endowed chair of Women in Science and Health (WISH) for her outstanding contributions in biomedical research.
(2 min. read)
A team at Children’s National performed the first treatment with sonodynamic therapy utilizing low intensity focused ultrasound (LIFU) and 5-aminolevulinic acid (5-ALA) medication on a pediatric patient. The treatment was done noninvasively through an intact skull.
(3 min. read)
In an editorial, Roberta L. DeBiasi, M.D., M.S., provided a comprehensive review of what is known about the harmful effects of SARS-CoV-2 infection in pregnant women themselves, the effects on their newborns, the negative impact on the placenta and what still is unknown amid the rapidly evolving field.
(2 min. read)
Doctors at Children’s National used a staged, hybrid cardiac surgical strategy to care for a patient who was born with hypoplastic left heart syndrome (HLHS) at 28-weeks-old. Hybrid heart procedures blend traditional surgery and a minimally invasive interventional, or catheter-based, procedure.
(4 min. read)
In a review article in Seminars in Pediatric Surgery, Marc Levitt, M.D., chief of the Division of Colorectal and Pelvic Reconstruction at Children’s National, discussed the history of pediatric colorectal and pelvic reconstructive surgery and described the key advances that have improved patients’ lives.
(11 min. read)


In this study, the authors at Shanghai Children’s Medical Center utilized next-generation sequencing (NGS) to analyze the data of patients with short stature to better understand the etiologies of short stature.
Andrew Dauber, M.D., M.M.Sc., division chief of Endocrinology at Children’s National Hospital, shared expert commentary on a recent study published in The Journal of Clinical Endocrinology & Metabolism that explores associated risk factors of short stature as identified by exome sequencing in children.
In this study, the authors at Shanghai Children’s Medical Center utilized next-generation sequencing (NGS) to analyze the data of patients with short stature to better understand the etiologies of short stature.
“This was a large-scale study looking at 814 children with short stature and at least one more clinical feature suggestive of a genetic condition who underwent comprehensive genetic testing at Shanghai Children’s Medical Center,” explains Dr. Dauber. In this study, the authors identified a potential genetic etiology in 361 of the patients, which is 44% of the cohort.
“It is important to note that the yield of genetic testing was highly variable depending on the clinical presentation of the child,” said Dr. Dauber. “For example, patients with associated congenital anomalies or a suspected skeletal dysplasia had a diagnostic yield of 56% and 65% respectively, while patients with isolated severe short stature (defined as a height below -3 SDS) only had a yield of 11%.”
Dr. Dauber noted that the overall high yield is reflective of the types of patients who are referred to this specialty center, and the expected yield in a more general pediatric setting is likely much lower.
“This study helps shed light on the prevalence of those patients in a large cohort of children presenting for evaluation of short stature,” shared Dr. Dauber. “I am hopeful that targeted treatments will improve growth in these children.”
While this study provides new insights into the underlying causes behind short stature in patients with differing phenotypes, the authors indicate that additional large-scale studies on short stature exome sequencing are warranted.
Dr. Dauber also pointed to the fact that the authors note a large number of the patients in this study had undiagnosed Rasopathies, such as Noonan syndrome. “There were also 31 patients with FGFR3 mutations, 6 patients found with ACAN (Aggrecan) mutations and 2 with NPR2 mutations,” said Dr. Dauber.
“At Children’s National, we are currently conducting a clinical trial of vosoritide, a novel growth promoting agent which targets the growth plate in children with selected genetic conditions including Noonan syndrome and patients with mutations in FGFR3, ACAN, and NPR2,” included Dr. Dauber. Preliminary results from this clinical trial were recently presented by Dr. Dauber at the Pediatric Endocrine Society annual meeting.
You can read the full study Clinical Profiles and Genetic Spectra of 814 Chinese Children With Short Stature in The Journal of Clinical Endocrinology & Metabolism.
Aggrecan (ACAN) is a large protein found in joint cartilage and growth plates.
Andrew Dauber, M.D., M.M.Sc., division chief of Endocrinology at Children’s National Hospital, and colleagues recently published two papers that describe the phenotypic spectrum of aggrecan deficiency and look at treating the condition with human growth hormone.
Aggrecan (ACAN) is a large protein found in joint cartilage and growth plates. It allows joints to move smoothly and without pain. Aggrecan deficiency — due to heterozygous mutations in the ACAN gene — causes dominantly inherited short stature and, in many patients, early-onset osteoarthritis and degenerative disc disease.
In 2017, Dr. Dauber led an international consortium of researchers that published a manuscript describing the phenotypic spectrum of 103 individuals – 70 adults and 33 children, including 57 females and 46 males – from 20 families with ACAN mutations. In the study, Dr. Dauber and his colleagues established that short stature and accelerated bone age is common among people with ACAN mutations.
In a new study published in the American Journal of Medical Genetics Part A, Dr. Dauber and colleagues further characterize the phenotypic spectrum of aggrecan deficiency, with an emphasis on musculoskeletal health.
Twenty-two individuals from nine families were enrolled in the study. Recorded histories and examinations focused on joint health, gait analysis, joint specific patient reported outcomes and imaging.
“We performed a detailed analysis of the musculoskeletal manifestations in patients with mutations in the aggrecan gene,” says Dr. Dauber. “We found that patients with mutations in this gene had significant short stature which worsened with age. There was a high prevalence of joint complaints and arthritis in adults, and we were able to detect pre-symptomatic joint damage in children using knee MRIs.”
Until now, it was unknown how to treat children with aggrecan deficiency. “Providing growth hormone therapy to children with ACAN gene mutations is relatively new in the field of pediatric endocrinology,” explains Dr. Dauber. “Previously, the assumption was that this was just short stature.”
In a new study, published in The Journal of Clinical Endocrinology and Metabolism, Dr. Dauber and colleagues reported the results of a trial that evaluated the efficacy and safety of recombinant human growth hormone (rhGH) therapy on linear growth in children with ACAN deficiency.
“This is the first prospective trial of growth hormone therapy in patients with mutations in the aggrecan gene,” says Dr. Dauber. “Mutations in the gene are the cause for short stature in approximately 2% of individuals with idiopathic short stature.”
The open-label, single-arm, prospective study enrolled ten treatment-naïve patients with a confirmed heterozygous mutation in ACAN. Participants were treated with rhGH (50 µg/kg/day) over 1 year. Main outcomes measured were height velocity and change in height standard deviation score.
The authors found that growth hormone led to short term improvements in growth rate over the course of the year. The treated patients had their growth rate increase from 5.2 centimeters per year to 8.3 centimeters per year while on therapy.
In 2019, the researchers received the 2019 Human Growth Award at the Pediatric Endocrine Society’s Annual Meeting for an abstract related to this work, entitled “Clinical Characterization and Trial of Growth Hormone in Patients with Aggrecan Deficiency: 6 Month Data.”


A new Phase 2 study at Children’s National will look at using the drug vosoritide to promote growth in children with growth disorders.
A child’s growth is often measured by pediatricians during routine physicals to identify abnormalities of growth and stature. An abnormality in these measurements could mean a child has a growth disorder. There are many different causes of growth disorders. Some can be the result of defects in genes related to the growth plate, which is the tissue near the end of long bones that grows as the child grows. Children with a growth disorder can present many different symptoms including short stature, joint pain, heart problems, bone problems and developmental delays. Scientists still have a lot to learn about what exactly causes these genetic growth disorders and treatments are limited, especially in the pediatric population. Growth hormone is not uniformly helpful and has only been approved for a small number of conditions.
Vosoritide is an investigational drug that directly targets the growth plate to promote growth. It is an analog of the amino acid C-type natriuretic peptide (CNP). It binds its receptor on healthy cartilage cells called chondrocytes and is currently under investigation in clinical trials as a treatment for the bone growth disorder achondroplasia.
“Vosoritide for Selected Genetic Causes of Short Stature” is a Phase 2 study currently open at Children’s National Hospital. This study will target five types of genetic short stature including SHOX deficiency, hypochondroplasia, Rasopathies (which includes Noonan syndrome), heterozygous NPR2 defects and CNP deficiency.
Thirty-five children with short stature will be enrolled and followed for a six-month observation period to obtain baseline growth velocity, safety profile and quality of life assessment. Study participants will then be treated with vosoritide for 12 months and will be assessed for safety and improvement in growth outcomes.
“Many patients who present with short stature likely have genetic defects in genes involved in growth plate physiology. Those patients with selected causes of short stature that interact with the CNP pathway may benefit from treatment with vosoritide, which directly targets the growth plate,” said Andrew Dauber, M.D., MMSc., lead investigator of this clinical study and chief of Endocrinology at Children’s National Hospital, a program ranked in the top 10 by U.S. News & World Report. “In this study, our goal is to understand if vosoritide may be a safe and effective treatment option for certain genetically defined short stature syndromes.”
This clinical trial has been approved by the FDA and funded by BioMarin. Children’s National is the only site in the world offering this therapy for patients with these conditions. The study is currently underway and subject recruitment is ongoing. There are 9 participants enrolled to date.
“This study could fundamentally change the way we treat certain growth disorders”, says Dr. Dauber.
For more information on the clinical trial, click here.


Growth hormone therapy is one of the most common treatments for short stature in children. However, endocrinologists report mixed outcomes, even when children have the same underlying condition as the cause of their short stature. Despite research into a variety of potential causes for this unpredictable response, there is still very little scientific evidence to help physicians predict whether children with short stature who are treated with growth hormone will respond to the treatment or not.
A study published in the Journal of Clinical Endocrinology and Metabolism took up this question with an eye to the genetic factors that might predict response by conducting the first ever genome-wide association study of response to growth hormone.
“Previous disease-specific models have been developed using multiple clinical variables such as parental height, age at treatment start, peak hormone levels and doses, and birth parameters, however, these clinical parameters only partially predict variation in response,” wrote the study authors, including Andrew Dauber, M.D., first author and chief of the division of Endocrinology at Children’s National Hospital. “Our goal was to perform a large-scale genome-wide study to provide a comprehensive assessment of how common genetic variation may play a role in growth hormone response.”
To accomplish this, the study combined five cohorts from across the world to identify 614 individuals for further review. All patients had short stature caused by conditions including growth hormone deficiency, small for gestational age, or idiopathic short stature (no previously identified cause), who received growth hormone as treatment.
Interestingly, the researchers found no overwhelming genetic predictors of response to growth hormone. They did identify a few signals that may potentially play a role in the body’s response to growth hormone but noted those signals will need further exploration. The study also ruled out the idea that genetic predictors of height in the general population might predict response to growth hormone.
“Identifying genetic predictors of how a child with short stature will respond to growth hormone would be an important step forward for clinicians and parents to make informed decisions about the right treatment approach,” says Dr. Dauber. “Though we didn’t find any specific indicators, this large-scale study has allowed us to rule out some previously held assumptions and offers several new avenues to explore.”
The study was conducted in collaboration with Pfizer and Boston Children’s Hospital.


M takes a photo with her daughter in Washington, where they learned they have an ACAN gene mutation that causes short stature.
On Sunday, April 28, 2019 a team of researchers received the 2019 Human Growth Award at the Pediatric Endocrine Society’s Annual Meeting for their abstract, entitled “Clinical Characterization and Trial of Growth Hormone in Patients with Aggrecan Deficiency: 6 Month Data,” and presented this at the PES Presidential Poster Session.
Eirene Alexadrou, M.D., a fellow at Cincinnati Children’s Hospital Medical Center, accepted the award and honorarium, while ongoing research is underway. This study started in 2017, with the objective of characterizing the phenotypic spectrum and response to a standardized regimen of growth hormone in a small cohort of 10 patients and their families.
In 2017, Andrew Dauber, M.D., MMSc., the division chief of endocrinology at Children’s National Health System, led an international consortium of researchers in publishing a manuscript describing the phenotypic spectrum of 103 individuals – 70 adults and 33 children, including 57 females and 46 males – from 20 families with aggrecan gene (ACAN) mutations.
Dr. Dauber and his colleagues have established that short stature and accelerated bone age is common among people with ACAN mutations. In a review of retrospective data, including patients treated with a variety of growth-promoting therapies at varying doses, the research team found that over the first one, two and three years of treatment, the standard deviation scores (SDS) for height increased by .4, .7 and 1, respectively. The current abstract now describes seven children enrolled in a prospective standardized trial of growth hormone therapy. After six months of treatment, the children have increased their height SDS by an average of 0.46.
Additionally, the researchers are performing an in-depth look at the joint effects, including special MRIs of the knees. They found that two of the children had a problem with their knee cartilage called osteochondritis dissecans. They had not yet presented with clinical symptoms. The researchers hope that early intervention with physical therapy can help prevent significant joint disease in the future.

M, her daughter, and M’s mother take a photo in Cincinnati, where they are participating in a clinical trial for aggrecan deficiency.
“Providing growth hormone therapy to children with ACAN gene mutations is relatively new in the field of pediatric endocrinology,” notes Dr. Dauber. “Previously, the assumption was that this was just short stature. We’ll continue to diagnose ACAN mutations in a clinical setting and work with families to reduce the risk of complications, such as joint problems or early-onset arthritis, which may co-occur with this gene mutation.”
As an example, Dr. Dauber met an 8-year-old patient several months ago who presented with symptoms of short stature. The patient is healthy, confident and still growing so her mother wasn’t worried about her but she made the appointment to see if there was an underlying cause to her daughter’s short stature. Her family history revealed clues to an ACAN mutation, which was later confirmed through genetic tests. Her mom, M, stands 4’8; her grandmother is 4’9. Her great grandmother was short and her great, great grandfather was 5’1. Short stature and joint problems run in the family. Once M mentioned she had osteochondritis dissecans and a hip replacement, she provided a textbook case study for carrying the ACAN mutation.
After the appointment, M shared the news with her mother about the possibility of having aggrecan deficiency. After taking genetic tests, M, her mother and M’s daughter learned they all have the ACAN mutation, and enrolled in the study that Dr. Dauber is guiding. Suddenly, it all made sense. After examining family photos, they traced the ACAN mutation back through four generations.
They could tell what relatives had an altered copy of the ACAN gene. M had it, while her two sisters did not. M’s mother was an only child, so she didn’t have aunts or uncles to compare her mother’s height to, but M’s grandmother was short, while her grandmother’s brother was average height. Although her mother’s family was from Germany, she learned that there is no specific ancestry associated with this mutation. It happens by chance and is passed down from a single parent to, on average, half of their children, a form of genetic inheritance called autosomal dominant transmission.

M’s great grandfather was noticeably shorter than her great grandmother, who was 5’4.
Through further research, M learned that the ACAN gene provides instructions for producing aggrecan protein, which is essential for bone growth, as well as for the stability of cartilage that lines bones and joints, explaining her recurring joint problems.
She also looked into the future, examining potential risk factors for her daughter: joint pain and bone conditions, which could contribute to arthritis, hip dysplasia and back problems.
The diagnosis now makes it easier for M and her daughter to favor bone-building activities that are easy on the joints, like swimming or water aerobics, instead of gymnastics and weight lifting. After having a hip replacement, M was careful to supplement with calcium and vitamin D. Now, she’ll take the same steps to ensure optimal bone health for her daughter. She’ll work with orthopedic specialists as her daughter grows into her pre-teen and adolescent years, carefully monitoring joint pain – altering activities that are tough on the joints, as necessary.
M let her daughter make a decision about growth hormone therapy, which she decided to try. The benefits of the treatment, increased height, carry inconveniences, such as taking daily shots, but they are sticking with it.
“We’re at the tip of the iceberg with research that explores this gene mutation,” says Dr. Dauber. “We’ll continue to study these families, and more, over time to assess growth patterns and gene expression, which may reveal other mutations associated with short stature or joint problems, and guide future treatment options. It was a coincidence that this family had the ACAN mutation and scheduled an appointment, while we’re conducting this study. Otherwise, they may not have had an answer since this is fairly new research.”
M and her daughter are happy to be part of this study, which they will participate in for the next few years. M’s mother is also glad to participate. She made a different choice, decades ago, to reject hormone treatment when it was offered to her for undiagnosed short stature, but she’s sharing genetic clues, which may influence treatment options for her granddaughter and for her family’s next generation.
The original study, “Clinical Characterization of Patients with Autosomal Dominant Short Stature due to Aggrecan Mutations,” appeared in the Feb. 2017 issue of the Journal of Clinical Endocrinology and Metabolism, and published as an online advance on Nov. 21, 2016.
Thirty-six researchers collaborated on this original paper, which was funded by 16 international health institutes and foundations, including the Eunice Kennedy Shriver National Institute of Child Health and Human Development at the National Institutes of Health, the Swedish Research Council, the Swedish Governmental Agency for Innovation Systems, the Marianne and Marcus Wallenberg Foundation, the Stockholm County Council, the Swedish Society of Medicine, Byggmastare Olle Engkvist’s Foundation, the Sao Paulo Research Foundation, the Spanish Ministry of Education and Science, the Czech Health Research Council and the Ministry of Health, Czech Republic.
An international collaboration resulted in the identification of a new cause of dwarfism: mutations in a gene known as DNMT3A.
Beyond diabetes, short stature is the most common reason for children in the U.S. to visit an endocrinologist. For the vast majority of children with short stature, the cause remains unknown – even though many of these conditions stem from an as-yet unidentified genetic cause, says Andrew Dauber, M.D., M.M.Sc., division chief of Endocrinology at Children’s National Health System.
“Parents are concerned about why their child isn’t growing and if there are other complications or health problems they’ll need to watch out for,” he says. “Without a diagnosis, it’s very hard to answer those questions.”
Dauber’s research focuses on using cutting-edge genetic techniques to unravel the minute differences in DNA that limit growth. This research recently led him and his colleagues to identify a new cause of dwarfism: mutations in a gene known as DNMT3A. The discovery, which the team published in the January 2019 Nature Genetics, didn’t happen in isolation – it required a rich collaboration of labs spread across the world in Scotland, Spain, France and New Zealand, in addition to Dauber’s lab in the U.S.
The journey that brought Dauber into this group effort got its start with a young patient in Spain. The boy, then four years old, was at less than 0.1 percentile on the growth curve for height with a very small head circumference and severe developmental delays. This condition, known as microcephalic dwarfism, is incredibly rare and could stem from one of several different genetic causes. But his doctors didn’t know the reason for this child’s specific syndrome.
To better understand this condition, Dauber used a technique known as whole exome sequencing, a method that sequences all the protein-coding regions in an individual’s entire genome. He found a mutation in DNMT3A – a change known as a de novo missense mutation, meaning that the mutation happened in a single letter of the boy’s genetic code in a way that hadn’t been inherited from his parents. But although this mutation was clear, its meaning wasn’t. The only clue that Dauber had as to DNMT3A’s function was that he’d read about overgrowth syndromes in which the function of this gene is lost, leading to large individuals with large heads, the exact opposite of this patient’s condition.
To gather more information, Dauber reached out to Andrew Jackson, Ph.D., a researcher who studies human genes for growth at the University of Edinburgh in Scotland. Coincidentally, Jackson had already started studying this gene after two patients with a shared mutation in a neighboring letter in the genetic code – who also had short stature and other related problems – were referred to him.
Dauber and his colleagues sent the results from their genetic analysis back across the Atlantic to Jackson’s Edinburgh lab, and the doctors from Spain sent more information to Jackson’s lab, including the patient’s clinical information, blood samples and skin biopsy samples. Then the whole team of collaborators from around the globe set to work to discover the processes influencing short stature in each of these three patients.
Their results showed that these mutations appear to cause a gain of function in DNMT3A. This gene codes for a type of enzyme known as a methyltransferase, which places methyl groups on other genes and on the protein spools called histones that DNA wraps around. Each of these functions changes how cells read the instructions encoded in DNA. While the mutations that cause the overgrowth syndromes appear to allow stem cells to keep dividing long past when they should taper off and differentiate into different cell types – both normal processes in development – the gain of function that appears to be happening in these three patients prompts the opposite situation: Stem cells that should be dividing for a long time during development stop dividing and differentiate earlier, leading to smaller individuals with far fewer cells overall.
The researchers confirmed their findings by inserting one of the gain-of-function human DNMT3A mutations into a mouse, leading to short animals with small heads.
Eventually, says Dauber, these findings could help lead to new treatments for this and other types of dwarfism that act on these genetic pathways and steer them toward normal growth. These and other scientific discoveries hinge on the type of international collaboration that he and his colleagues engaged in here, he adds – particularly for the types of rare genetic syndromes that affect the patients that he and his colleagues study. With only a handful of individuals carrying mutations in certain genes, it’s increasingly necessary to combine the power of many labs to better understand the effects of these differences and how doctors might eventually intervene.
“The expertise for all aspects of any single research project is rarely centered in one institution, one city, or even one country,” Dauber says. “Often, you really need to reach out to people with different areas of expertise around the world to make these types of new discoveries that can have pivotal impacts on human health.”


Andrew Dauber, M.D., hosts an AMA chat with Reddit’s science community and offers feedback about height, growth disorders and pediatric endocrinology.
Andrew Dauber, M.D., MMSc., the division chief of endocrinology at Children’s National, spoke about epigenetics – how genes are expressed – and about all things related to pediatric endocrinology in a recent Ask Me Anything (AMA) chat with Reddit’s science community.
We’ve selected highlights from several questions Dr. Dauber received. You can view the full AMA discussion on Reddit.
Q1: What will the future of type 1 diabetes treatment look like?
As a pediatric endocrinologist, Dr. Dauber sees a lot of patients with type 1 diabetes. He predicts technology will pave the way for advancements with continuous glucose monitoring and encourage a ‘real-time’ interaction between patients and providers:
“I anticipate that within a few years, everyone will have access to continuous glucose monitoring technology and that these will be seamlessly connected to insulin pumps or artificial pancreas technologies,” types Dr. Dauber in response to the first AMA question. “I also think there will be more virtual interaction between medical providers and patients with doctors and nurses reviewing blood sugar data in the cloud.”
Q2: What height range is considered normal for a growing child? What is the difference between short stature and a height problem?
The Centers for Disease Control and Prevention has a growth chart, which shows ‘normal’ ranges, based on statistical definitions of height in the general population.
“The truth is that I know plenty of people who have heights below the ‘normal’ population, and they don’t think they have a problem at all,” says Dr. Dauber. “From a genetics point of view, the question can be reframed: When do we call a genetic variant a ‘mutation’ versus a rare variant in the population? For example: If there is a genetic change that 1 in a 1,000 people have that causes you to be 2 inches shorter – is that a problem? Is that a disease?”
“From a clinical perspective, I tend to have a discussion with my patients and their families and ask them how their stature is affecting their lives and whether changing that would really make a meaningful difference,” adds Dr. Dauber. “I believe that this is a very personal decision but people need to be realistic about expected outcomes.”
Q3: What are your favorite case studies about atypical growth or height patterns?
Dr. Dauber references two case studies about growth and puberty:
The growth case study refers to the PAPPA2 gene, which was particularly meaningful for Dr. Dauber since he got to know the family and was able to provide answers to a previously undiagnosed medical mystery about short stature. This research is also opening future studies and analysis about the regulation of IGF-1 bioavailability.
The puberty case study looks at the opposite end of growth and development: precocious puberty. In this case an inherited MKRN3 gene mutation resulted in new insight about the regulation of pubertal timing: Deficiency of MKRN3 caused central precocious puberty in humans. Girls who had inherited the mutated genes from their father (an imprint gene) started to develop breasts before age 6. The results were published in The New England Journal of Medicine.
Q4: What are the differences with consistent and inconsistent growth disorders? Could one arm or leg experience accelerated or stunted growth?
“Most genetic disorders that affect growth will have a uniform effect throughout the body as they are likely to affect all aspects of the skeleton,” says Dr. Dauber. “That being said, there are some notable exceptions such as Russell-Silver syndrome which presents with body asymmetry. There are also somatic mutations (mutations which are just present in some cells in the body) that can lead to segmental areas of overgrowth leading to asymmetry.”
Q5: Can you predict height and growth by looking at genetic factors? What are your thoughts about polygenic risk scores?
“Polygenic risk scores will probably play more of a role in the future to help determine risk of a certain disease,” says Dr. Dauber. “Right now, for most conditions, the risk score does not explain a substantial enough fraction of the variation to help with prediction.”
Dr. Dauber discusses how this works for height, a highly hereditable trait, in The Journal for Clinical Endocrinology and Metabolism. In the review, Dr. Dauber and the study co-authors note that individuals with extreme heights are more likely to have abnormal stature as a result of a severe mutation that causes a growth disorder. For these individuals, whole exome sequencing may reveal gene mutations.
However, the study authors note that for now, the role of these technologies in individuals with extreme stature but without any syndromic features has not been rigorously and systematically explored. (Dr. Dauber and a team of endocrinologists from leading children’s hospitals are currently using electronic health records to study and track these types of genetic clues over time.)
Q6: The general public is excited about genetics and ongoing research, especially with consumer applications – such as genetic tests, including 23andMe. What misconceptions about genetics do people have? What ethical concerns do geneticists share right now?
“Many people think that genetics is completely deterministic,” says Dr. Dauber. “In reality, most genetic variants influence a person’s predisposition toward a trait or disease but don’t actually determine the outcome. Also, the genetic sequence itself is just the first step. Epigenetics, gene regulation, and gene-environment interactions are all important and we are just scratching the surface of understanding these areas.”
“I think that people engaged in genetics research are very interested in the ethical questions,” adds Dr. Dauber. “The problem is that technology is advancing at such a rapid pace, that often consumers are using technologies in ways that we haven’t yet had time to figure out the ethics for. The medical community is often playing catch up.”
Q7: Aside from using gene modifications to cure diseases, where or when should we draw the line in terms of enhancement?
“I think genetic modification for enhancement is a very dangerous slippery slope that we should avoid,” says Dr. Dauber. “We really don’t know the full effect of many genes and by enhancing them, we could be causing lots of problems that we can’t anticipate. There is a reason that evolution is a slow process that happens over millions of years. I think we need to start with the most devastating diseases and try to cure those first.”
Q8: Would it be ethical to use CRISPR on the genes for short stature to produce tall offspring if the risks are sufficiently small? This would be similar to what Dr. He did, but without the ethical violations.
“This is a fascinating question and it will become more of an issue over time,” says Dr. Dauber. “Where do we draw the line between fixing, preventing disease and enhancing physical function? Personally, I think using genome editing to promote height is a terrible idea. Our current perception that taller height is more desirable is a social construct and varies by culture. This idea also changes over time.”
Q9: Overall, how does this fit into meeting unmet medical needs?
“I would be very wary about trying to design our children’s physical features,” Dr. Dauber notes. “We need to figure out as a society what diseases are sufficiently problematic that we feel comfortable trying to eliminate them via genome editing.”
Q10: How many genes control acromegaly? Is it possible (in theory) to Top of Formselect them just to gain the positive effects of gigantism without the health risks?
Dr. Dauber explains that acromegaly, a condition often referred to as gigantism, is caused by a growth hormone-producing tumor. There are a few genes known to cause these tumors, including the AIP, and there was recently a genetic cause of X-linked gigantism, which was published in The New England Journal of Medicine.
“This basic idea is a good one,” notes Dr. Dauber. “We can find genes that when mutated can cause tall stature – and then try to manipulate those pathways. A great example is the NPR2 gene, which when mutated can cause short or tall stature. This pathway is being targeted for therapeutics related to achondroplasia.”
The National Institutes of Health (NIH) refers to achondroplasia as ‘short-limbed dwarfism,’ which results in an average-sized trunk with short limbs, especially arms and legs, due to a lack of cartilage turning into bone. The average height of an adult male with achondroplasia is 4 feet, 4 inches, while the average height of adult females with achondroplasia is less than 4 feet, 1 inch. In this case, manipulating growth pathways may help alleviate health problems associated with achondroplasia: lack of mobility or range of motion, an enlarged head, apnea, ear infections and spinal stenosis, or a compression or pinching of the spinal cord.
Q11: Give us a history lesson. Why are there variations of height within populations, such as Asia and Latin America?
“The average height in a population is due to the influence of literally thousands of common genetic variants,” says Dr. Dauber. “These population differences have evolved over thousands of years due to a combination of migration and selection. There is a well-known difference in the genetic makeup of various populations which likely underlies the differences across the globe. There are even differences within Europe.”
Q12: Are there examples of pseudoscience or theories about growth, such as recommendations to eat a certain food instead of taking growth hormones to correct for a growth disorder, which runs contrary to scientific evidence, that drive you crazy?
“I don’t really get bothered by crazy theories, but it is upsetting when patients and their families get swindled into spending their money on therapies that aren’t truly effective,” says Dr. Dauber. “People ask me all the time if a certain food or exercise can make their child taller. The bottom line is that in a well-nourished (and healthy) child, there is no magical food that is going to make them tall.”
Q13: According to almost every theory of how life evolved on Earth, from religion to evolution, we all have one common ancestor. In theory doesn’t that make us all cousins?
“Yes, just very distant ones,” says Dr. Dauber. “People always point out the vast number of differences between races but in fact we are all more than 99.9 percent identical on a genetic level.”
Stay on top of the latest pediatric endocrinology news by following @EndoDocDauber and @ChildrensHealth on Twitter.
What would happen if you suddenly stopped growing at age 12 or 13?
Solving genetic growth mysteries and scheduling regular appointments with pediatric endocrinologists is atypical for most parents and pediatricians.
However, for children with growth disorders – a classification that typically describes children below the third or above the 97th percentile of growth charts for their age – receiving a diagnosis is half the battle to reaching average height. Understanding and creating treatment for a growth disorder, which could stem from an underlying medical illness, a genetic mutation or a problem with endocrine function, such as the production or action of growth hormone, is often the next step.
For Andrew Dauber, M.D., MMSc., the chief of endocrinology at Children’s National Health System, a third step is to use these clues to create larger datasets and blueprints to identify risk factors for rare growth disorders. By understanding genetic markers of growth disorders, endocrinologists can identify solutions and create plans for multidisciplinary care to help children reach developmental milestones and receive coordinated care throughout their lifespan.
A case study that Dauber and his research team continue to explore is how to correct for mutations in the PAPPA2 gene, which regulates human growth by releasing a key growth factor called insulin-like growth factor 1 (IGF-1). Dauber and his colleagues recently described a mutation in PAPPA2, observed in two families with multiple children affected with significant short stature. He found that this mutation decreased the bioavailability of IGF-1, stunting the growth and development of the children who carry this mutation.
While the PAPPA2 mutation is rare, endocrinologists, like Dauber, who understand its function and dysregulation can create solutions to support IGF-1 bioavailability, thereby supporting healthy growth and development in children.
Understanding barriers to IGF-1 function can also help researchers gain insight into the relationship between PAPPA2, levels of circulating insulin in the body, which could cause insulin resistance, and other growth hormones. For now, Dauber and his research team are exploring how to use PAPPA2 to increase IGF-1 in circulation among people with height disorders in the hopes of improving their growth.
“The population of children who have PAPPA2 mutations is small and we’re finding out that two children could respond to the same treatment in different ways,” says Dauber. “One medication could work modestly in one child and support short growth spurts, such as growing by 5 or 6 cm a year. It could also create undesirable side effects, such as headaches and migraines in another, and render it ineffective. However, the clues we walk away with enable us to test new solutions, and confirm or dissolve our hunches, about what may be preventing the bioactive release of essential growth hormones.”
To generate controls for healthy patterns of growth and development, Dauber and his research team are analyzing the relationship between PAPPA2, STC2 and IGFBP-3 concentrations among 838 relatively healthy pediatric participants, ages 3-18, with traditional growth patterns.
They are studying PAPPA2, STC2 and intact IGFBP-3 concentrations throughout childhood and the researchers are already surprised to find PAPPA2, a positive modulator of growth and IGF- bioavailability, decreased with age, while STC2, a negative modulator and traditional growth inhibitor, increased with age.
“As pediatric endocrinology researchers and clinicians, we’re looking at the pathology of traditional growth patterns and growth disorders with an open mind,” says Dr. Dauber. “These data sets are invaluable as they confirm or challenge our theories, which enable us to create and test new forms of personalized treatments. We’ll continue to share this knowledge, which informs other researchers and accelerates the field of pediatric endocrinology.”
This research was presented at the annual meeting of the European Society of Pediatric Endocrinology in Athens on Sept. 28, 2018.
Dauber and his research team will present their findings at endocrinology conferences and grand rounds throughout 2018 and 2019.
To view Dr. Dauber’s most recent research and pediatric endocrinology reviews, visit PubMed.
