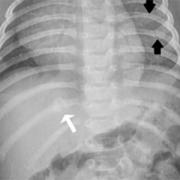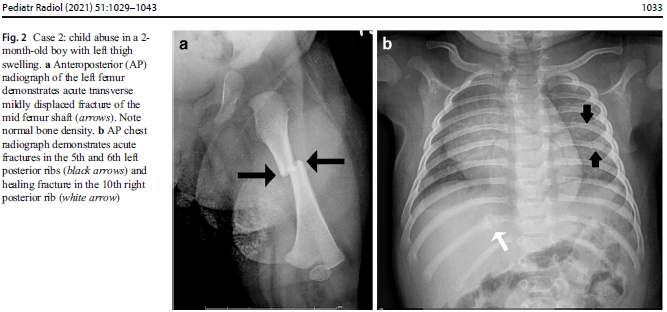
In suspected child abuse cases, pediatric specialists are often called for clinical consultations or subpoenaed to courtrooms to discuss unexplained fractures and hemorrhage. During routine clinical practice, Children’s National Hospital (CNH) geneticists, child abuse pediatricians, orthopedists and radiologists use a multidisciplinary, stepwise approach that differentiates genetic disorders from cases of suspected child abuse. These teams provide a clear process for when clinical and radiological review suffices versus cases when judicious use of genetic testing and biochemical testing should be considered.
Every year, approximately 675,000 children suffer abuse or neglect, and at least 1,700 die from abuse — one of the leading causes of childhood morbidity and mortality nationwide.
Ill-intended arguments regarding genetic disorders often reach the court. Out of the 7,000 known rare diseases, rare bone diseases constitute about 5% of the cases. When there are true genetic disorders like osteogenesis imperfecta (OI), a group of genetic disorders that cause fractures, orthopedic specialists help tremendously with diagnosis and treatment. The Children’s National Bone Health Program specializes in caring for healthy children, children with genetic bone conditions, and children whose bones have been damaged from illness or poor nutrition. Their team of experts enhances treatment to meet the needs of each child help us provide the best possible care for children with a broad range of bone health conditions When a rare condition that causes bone fragility is suspected, these teams work together to provide proper diagnosis and management.
“OI is a diagnosis that can be made clinically with the help of geneticists, radiologists and orthopedists,” said Tanya Hinds, M.D., a child abuse pediatrician at Children’s National. “Outside of the newborn period, multiple unexplained fractures in infants with radiologically normal bones is suspicious for child physical abuse, not OI.”
When these regional cases reach the courtroom, Children’s National pediatricians often serve as clinician-educators and expert witnesses. According to Children’s National experts, clinicians must share the best available medical practices in both the hospital and courtroom. Unfortunately, in some cases around the country, a handful of expert medical witnesses provide unique and unsubstantiated opinions, sometimes claiming the presence of a rare genetic disorder as a cause of fracture or hemorrhage, when this has not been diagnosed by mainstream genetics specialists.
“On the part of expert witnesses, scientifically sound explanations versus unfounded hypotheses can influence outcomes in civil proceedings, which determine a child’s placement and criminal proceedings, which determine judgment on the perpetrators,” said Natasha Shur, M.D., medical geneticist at Children’s National, and Nathaniel Robin, M.D., professor and clinical genetics director at the University of Alabama in an editorial published in Current Opinion in Pediatrics.
Dr. Hinds works on behalf of children to provide the best and most comprehensive work-up in cases of unexplained fractures or hemorrhage. As a board-certified child abuse pediatrician, she is responsible for implementing the evidence-based practice guidelines of the American Academy of Pediatrics and other similar societies. Dr. Hinds mentioned that it is possible to use medical history, physical examination and diagnostic testing to differentiate traumatic causes of fractures and subdural hematomas from genetic causes, a belief she states is held by the vast majority of child serving clinicians.
“In cases of suspected child abuse, a multidisciplinary group of clinicians at Children’s National routinely provide comprehensive and top-rate care and consider alternative explanations for fractures,” said Eglal Shalaby-Rana, M.D., a radiologist at Children’s National who has partnered with the hospital’s Child and Adolescent Protection Center team on these challenging cases since 1991. “A multidisciplinary team is crucial to the evaluation and often includes additional specialists such as pediatric radiology, trauma surgery, hematology and in some cases genetics.”
Further, these clinician-educators and researchers at Children’s National call for increased publication and use of consensus guidelines such as the consensus statement on abusive head trauma published in Pediatric Radiology in 2018. “Consensus guidelines synthesize the best available medical evidence and should be the basis for both clinical practice and education offered in the courtroom,” said Drs. Shur, Hinds and Shalaby-Rana.
Distinguishing child abuse from genetic disorders
Drs. Hinds, Shalaby-Rana and Shur have served as expert witnesses and in turn wanted to come together to help develop frameworks that share scientifically sound information with peers who might encounter spurious arguments in courtrooms regarding genetic disorders as an explanation for physical abuse and inflicted fractures. Their 2021 literature review, published in Current Opinion in Pediatrics, addresses some of these issues.
To help distinguish child physical abuse from a genetic disorder, Drs. Shur, Hinds and Shalaby-Rana worked as a multidisciplinary team to highlight best practices in six instances when genetic disorders were raised as explanations for inflicted fractures or hemorrhage, including Elhers-Danlos syndrome (EDS), osteogenesis imperfecta (OI), Menkes Disease and Glutaric Acidemia type I. In some cases, these explanations could be reasonable but should be diagnosed using routine clinical and radiological review, and when indicated, genetic and biochemical testing.
For instance, EDS is a diagnosis that is sometimes erroneously used to explain multiple fractures in cases of suspected physical abuse and can be misused in courtrooms. The most common EDS type is hypermobile EDS, often found in late adolescence or early adulthood. In some cases, babies erroneously receive hypermobility exams, or clinicians perform hypermobility assessments on parents of children with unexplained fractures — neither practice is indicated. Instead, the Children’s National team points out that children should receive a medical evaluation using the standard guidelines set by the American Academic of Pediatrics, American College of Radiology and other professional societies.
Dr. Shur also collaborated with radiologists at Boston Children’s Hospital in a related review published in Pediatric Radiology. There, George et al. addressed the clinical and molecular diagnosis criteria for EDS to help radiologists prevent misdiagnosis and support clinicians when seeing patients with multiple fractures.
“It is disturbing that the unsubstantiated EDS infant bone fragility hypothesis continues to be advanced in civil and criminal child abuse proceedings when fractures are not part of the diagnosis criteria for EDS,” said George et al.
The clinicians noted that the Beighton score, which helps diagnose hypermobile EDS, is not intended for children younger than 8 years old. Additionally, since the score provides insufficient data, other EDS features must be present, such as skin findings and connective tissue abnormalities.
OI, known as ‘‘brittle bone disease,’’ is a group of disorders that rarely present only with unexplained fractures. The researchers emphasize that infants and children with mild OI do not present exclusively with multiple fractures, which are specific to physical abuse such as multiple, bilateral rib fractures and classic metaphyseal lesions. Drs. Shur, Hinds and Shalaby-Rana share that pediatric specialists could overcome the diagnostic challenges between OI and child abuse through the inclusion of a genetic team in some cases, during the medical evaluation while also considering various criteria, such as family history, physical examination and laboratory findings. Molecular testing may be required in some instances, but it cannot substitute traditional clinical and radiology evaluations, according to these clinician-researchers.
Similarly, while Menkes disease can present with intracranial hemorrhage and fractures like child physical abuse, there are other distinguishing characteristics unique to Menkes disease, such as hair and facial dysmorphism. In a third related case-review published in Pediatric Radiology, Shur, Hinds and Shalaby-Rana et al. emphasize that diagnostic difficulties may arises when a multidisciplinary evaluation is omitted. They call upon all clinicians to provide ethical testimony in civil or criminal proceedings and to continue to utilize a multidisciplinary approach during daily clinical practice.
Irresponsible testimony and predatory journals
According to George et al., in collaboration with Dr. Shur, there are flawed publications on EDS associated with infant bone fragility that do not follow the gold standard of the scientific community. They believe this hypothesis must be rejected by experts in the field of pediatric imaging to safeguard the scientific integrity of the discipline. The lack of scientific design, peer review process and transparency causes negative consequences in the courtroom and threatens the proper adjudication of cases of suspected child physical abuse.
“Irresponsible testimony increasingly enters medico-legal proceedings dealing with allegations of child abuse, and so-called expert witnesses regularly cite these deeply flawed publications — in addition to misquoting the medical literature, loosely interpreting medical findings, presenting fictitious findings, and excluding salient and widely accepted facts from consideration,” said George et al.
In these pieces of literature, our Children’s National multidisciplinary team members reviewed the best available evidence and their collective decades of patient experience to highlight standard processes, which differentiate child physical abuse as a cause of fractures and hemorrhage from rare disorders. Rare does not mean mysterious, and with education and a multidisciplinary approach, every child can receive the best possible medical work-up and care, according to Drs. Shur, Hinds and Shalaby-Rana. They urge all physicians to share only mainstream clinical medicine in the courtroom to help ensure the best possible social outcomes for children and their families.



























 A
A