Using targeted signaling pathway therapy to prevent pediatric glioma formation
Researchers at Children’s National Hospital identified a vulnerability in a developmental signaling pathway that can be hijacked to drive pediatric low-grade glioma (pLGG) formation, according to a pre-clinical study published in Developmental Cell. The study demonstrated that targeted treatment prevents tumor formation, long before irreversible damage to the optic nerve can cause permanent loss of vision. This finding will inform chemo-prevention therapeutic trials in the future.
Brain tumors are the most common solid tumors in children, the most prevalent of which are pLGGs. Approximately 10% to 15% of pLGGs arise in patients with the familial cancer predisposition syndrome known as neurofibromatosis type 1 (NF1). This is a genetic condition that increases risks of developing tumors along the nerves and in the brain.
Nearly 20% of children with NF1 develop pLGGs along the optic pathway, also known as NF1-associated optic pathway glioma (NF1-OPG). Despite many advances in cancer therapy, there are no definitive therapies available that prevent or alleviate the neurological deficits (i.e. vision loss) and that could improve the quality of life.
“The evidence presented can inform chemoprevention therapeutic trials for children with NF1-OPG,” said Yuan Zhu, Ph.D., scientific director and Gilbert Family Endowed professor at the Gilbert Family Neurofibromatosis Institute and associate director of the Center for Cancer and Immunology Research, both part of Children’s National. “This therapeutic strategy may also be applicable to children with the developmental disorders that are at high risk of developing pediatric tumors, such as other RASopathies.”
The mechanism of vulnerability to pLGGs during development is not fully understood. It has been implied that the cell population of origin for this debilitating tumor is transiently proliferative during development. The NF1 gene produces a protein that helps regulate normal cell proliferation, survival and differentiation by inhibiting MEK/ERK signaling. When there is loss of function in NF1, it abnormally activates the MEK/ERK signaling pathway and leads to tumor formation.
Certain cells that exist transiently during the normal development of the brain and optic nerve are vulnerable to tumor formation because they depend on the MEK/ERK signaling. In this study, researchers in Zhu’s lab identified cells that were MEK/ERK pathway dependent and grew during a transient developmental window as the lineage-of-origin for NF1-OPG in the optic nerve. The researchers used a genetically engineered pre-clinical model to design a transient, low-dose chemo-preventative strategy, which prevented these tumors entirely.
“When we provided a dose-dependent inhibition of MEK/ERK signaling, it rescued the emergence and increase of brain lipid binding protein-expressing (BLBP+) migrating GPs glial progenitors, preventing NF1-OPG formation,” wrote Jecrois et al. “Equally importantly, the degree of ERK inhibition required for preventing NF1-OPG formation also greatly improved the health and survival of the NF1-deficient model.”
Ongoing clinical trials using MEK inhibitors (MEKi) are being performed for children as young as 1 month old. Thus, it becomes increasingly feasible to design a chemo-preventative trial using a MEKi to treat children with NF1. These treatment paradigms may have the potential to not only prevent OPG formation, but also other NF1-associated and RASopathies-associated developmental defects and tumors.
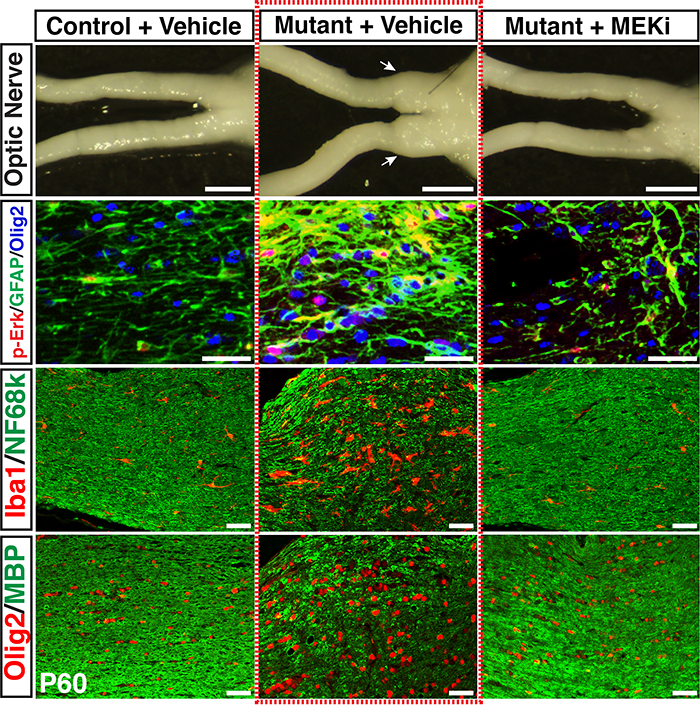
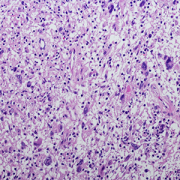
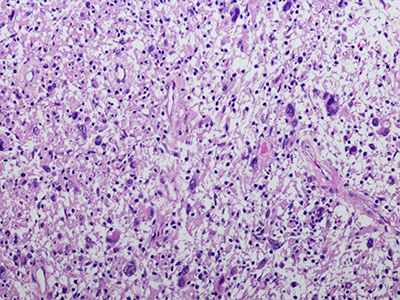
A transient low-dose MEKi treatment in a pre-clinical model prevents NF1-OPG formation. The middle panels highlighted by a red dashed box show an OPG in the optic nerve (arrows, top), exhibiting abnormal triply-labeled tumor cells, inflammation and nerve damage (the bottom three panels), which are absent in the normal (left panels) or MEKi-treated Nf1-deficient optic nerves (right panels). [Credit: Jecrois et al., Developmental Cell, (2021)]