The microscopic world of cell healing: A window into future therapies
 Unraveling how cells mend after injury serves as a key to unlocking potential therapies. Recent findings from the Center for Genetic Medicine Research at Children’s National Hospital offered surprising insights into the cell’s healing mechanisms by illuminating the intricate cellular responses to various types of injuries.
Unraveling how cells mend after injury serves as a key to unlocking potential therapies. Recent findings from the Center for Genetic Medicine Research at Children’s National Hospital offered surprising insights into the cell’s healing mechanisms by illuminating the intricate cellular responses to various types of injuries.
The study, featured on the back cover of the latest issue of Advanced Science, found that cells respond in distinct ways depending on the type of injury, such as a traumatic muscle tear that creates a large injury or tiny holes in the cell membrane caused by pathogenic proteins. Daniel Bittel, DPT, Ph.D., a research postdoctoral fellow at the Center for Genetic Medicine Research, said that cells are routinely injured from even everyday activities, such as walking up a flight of stairs.
“Injuries often involve damage to the plasma membrane,” Bittel said. “We wanted to investigate how healing happens at the subcellular level to better understand diseases and develop targeted therapies. We were especially curious about muscle cells because, interestingly, healthy ones get stronger the more that they are injured.”
The fine print
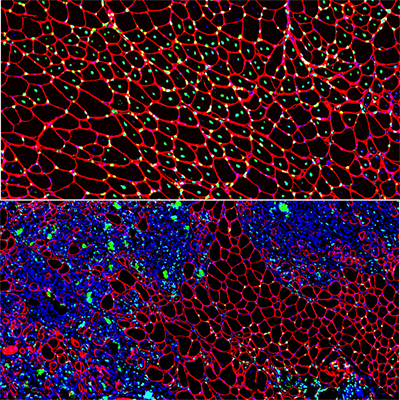
Using the center’s unique, custom-built microscope, the research team zoomed in on the process of cellular healing to watch how cells activate repair after injuries. Using a laser to damage the plasma membrane, they mimicked mechanically induced trauma. They also used a pathogen-derived protein to create nanoscale pinprick injuries in a cell’s plasma membrane that resemble those that are seen after strenuous muscle exertion.
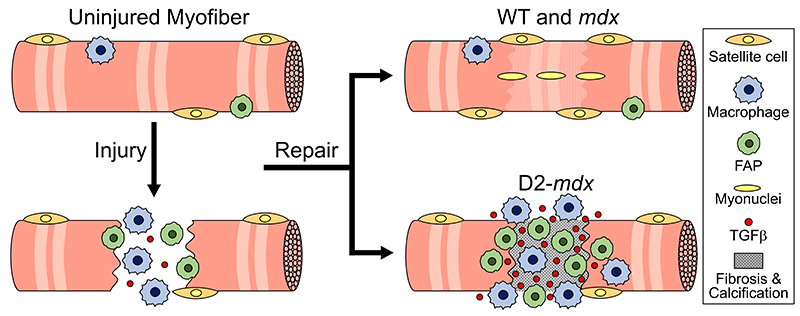
Then, they watched as cells went to work within seconds, engaging healing mechanisms tailored to the type of injury. In the case of a cell facing numerous pinpricks along the cell membrane, it immediately deployed the endocytic pathway used by the cells to eat and drink. This process helped remove the injurious agents and the tiny holes they made. However, with a larger mechanical injury, the cells demonstrated patience, allowing the plasma membrane to seal before clearing up the damage by the same endocytic pathway.
The big picture
The paper is part of an ongoing body of research on cell injury that will inform future investigations into a wide range of pediatric health issues including muscular dystrophies, injuries to neurons, orthopedic injuries from sports and other mechanical damage to tissues.
Jyoti Jaiswal, M.Sc., Ph.D., senior investigator at the Center for Genetic Medicine Research, said this work is foundational in the development of new therapies. “Knowing where the problem lies will help us figure out what therapy will work best and target the therapy to address the specific deficit,” he said. “This work will pave the way to help tailor therapies and tackle diseases more effectively.”