Given the wide variety of potential causes and seizure types, diagnosing pediatric epilepsy can be complex.
Pediatric epilepsy affects approximately 0.5-1% of children, with an increased prevalence in those with developmental disabilities. Epilepsy’s pathophysiology involves a range of factors including genetic mutations, structural brain abnormalities and metabolic disorders. Given the wide variety of potential causes and seizure types, diagnosing pediatric epilepsy can be complex.
Previously, finding the cause of pediatric epilepsy often involved laborious, piecemeal metabolic and chemical tests, making the process complicated for both patients and families. Technological advancements over the past decade, such as next-generation sequencing, have made genetic testing a vital tool in diagnosing and managing the disease through personalized treatment, leading to better outcomes.
In recent discussions, John Schreiber, M.D., explores the impact of genetic testing on pediatric epilepsy, highlighting its benefits, challenges and evolving role in treatment. Dr. Schreiber serves as Associate Chief of Epilepsy and Electroencephalography Operations, Medical Director of Electroencephalography and Medical Director of the Epilepsy Genetics Program at Children’s National Hospital.
“Genetic testing is becoming easier and less expensive to perform in patients with unexplained epilepsies, and thankfully now is being used much earlier in a patient’s epilepsy course,” Dr. Schreiber says. “It’s important to try to make that diagnosis early so we can ensure patients have the appropriate interventions early and avoid unnecessary tests and things that they don’t need.”
You can watch the full dialogue, “The Role of Genetic Testing on the Management Pediatric Epilepsy” on NeurologyLive.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2025/01/brain-with-brainwaves-feature.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2025-01-02 10:42:482025-01-07 10:40:05In the news: The role of genetic testing in pediatric epilepsy diagnosis and treatment
Dr. Eric Vilain accompanied by a fellow researcher at the new Research & Innovation Campus.
Children’s National Hospital announces a $12.8 million award from the National Institutes of Health’s National Human Genome Research Institute (NHGRI) to establish the only Pediatric Mendelian Genomics Research Center (PMGRC) as part of a new Mendelian Genomics Research Consortium. Researchers at Children’s National and Invitae — a leading medical genetics company — will identify novel causes of rare inherited diseases, investigate the mechanisms of undiagnosed conditions, enhance data sharing, and generally interrogate Mendelian phenotypes, which are conditions that run in families.
“Our overall approach provides an efficient and direct path for pediatric patients affected with undiagnosed inherited conditions through a combination of innovative approaches, allowing individuals, families and health care providers to improve the management of the disease,” says Eric Vilain, M.D., Ph.D., director of the Center for Genetic Medicine Research at Children’s National.
To accelerate gene discovery for Mendelian phenotypes and the clinical implementation of diagnosis, the consortium will leverage the broad pediatric clinical and research expertise of the Children’s National Research Institute and laboratories in partnership with Invitae. The Molecular Diagnostics Laboratory at Children’s National will provide genetic testing for patients in the Washington, D.C., metropolitan area. Invitae will provide genetic testing for patients from elsewhere in the U.S., giving the project a national reach and allowing researchers to leverage more robust data. Integrative analyses will be performed jointly with scientists at Children’s National and Invitae.
“Some patients have genetic test results that are ‘negative,’ meaning the results do not explain their condition. When a patient receives a negative result, it is challenging for parents and doctors to know what to do next,” says Meghan Delaney, D.O., M.P.H., chief of the Division of Pathology and Laboratory Medicine and Molecular Diagnostics Laboratory at Children’s National. “The project will provide an avenue to possibly find an explanation of their child’s condition. Besides filling an important clinical gap, the results will add new knowledge for future patients and the scientific community.”
“Too often parents of children suffering from a rare condition find themselves in a protracted diagnostic odyssey when early intervention could mean better overall outcomes,” says Robert Nussbaum, M.D., chief medical officer of Invitae. “We are proud to partner with Children’s National Research Institute on this important effort to identify the genetic cause of these rare conditions earlier and improve the chances that children with such conditions can receive the appropriate treatments and live healthier lives.”
Deciphering Mendelian conditions will help diagnose more of the estimated 7,000 rare inherited diseases and predict the tremendous variability of clinical presentations in both rare and common conditions caused by the same gene.
There is also a need to establish a new standard of care to bridge the gap in the use of genomic information from diagnosis to improved outcomes. The consortium will establish best practices for obtaining a genetic diagnosis, offering an explanation for the condition to affected patients, and is likely to provide additional explanations for basic biological mechanisms, increasing the knowledge of physiopathology and possibly leading to better condition management.
The PMGRC will enroll an average of 2,600 participants per year with suspected Mendelian phenotypes and previously non-diagnostic tests and their family members. The integration of multiple genomic technologies, including short and long read genome sequencing, optical genome mapping and RNA-sequencing, will enable these discoveries. To disambiguate uncertain variants and candidate genes, the PMGRC will use whole transcriptome analysis, RNA-sequencing, CRE-sequencing and functional modeling.
Since many Mendelian conditions first appear prenatally or during infancy, Children’s National will have a unique bed-to-bench-to-bed symbiosis. Patients eligible for the study will come from across the multiple specialty divisions of Children’s National, including the Children’s National Rare Disease Institute, and nationally through the partnership with Invitae. From there, experts from the Children’s National Center for Genetic Medicine Research will enroll patients and integrate the initial clinical test results with broad-based genomic interrogation, leading to new diagnoses and novel discoveries. Finally, the results will be verified and returned to clinicians, which will help inform targeted therapies.
Typically, the patients eligible for this study jump from specialist to specialist without an answer, have a condition that appears in other family members or they have symptoms involving more than one affected organ, which suggests a complex developmental condition. The PMGRC at Children’s National will help find answers to the causes of many puzzling pediatric conditions, providing faster clinical diagnoses and opening up pathways to potentially better treatments.
Dr. Vilain’s work will be based at the Children’s National Research & Innovation Campus on the grounds of the former Walter Reed Army Medical Center in Washington, D.C. The campus is also home to the Children’s National Rare Disease institute — one of the largest clinical genetics program in the United State that provides care to more than 8,500 rare disease patients.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2021/07/Dr.-Eric-Vilain-in-lab.png300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2021-07-15 10:00:272024-06-12 16:53:59Children’s National Hospital joins the Mendelian Genomics Research Consortium, receiving $12.8 million
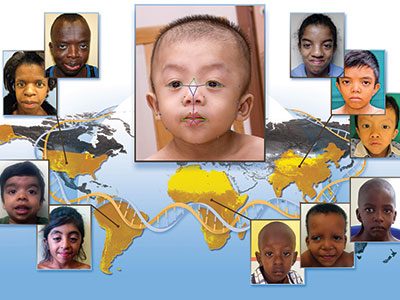
Children’s National Hospital has entered into a licensing agreement with MGeneRx Inc. for its patented pediatric medical device technology using objective digital biometric analysis software for the early and non-invasive screening of dysmorphic genetic diseases such as Noonan syndrome.
Children’s National Hospital has entered into a licensing agreement with life sciences technology company MGeneRx Inc. for its patented pediatric medical device technology using objective digital biometric analysis software for the early and non-invasive screening of dysmorphic genetic diseases. The technology, developed by a multidisciplinary Children’s National team led by Marius George Linguraru, D.Phil, M.A., M.Sc., of the Sheikh Zayed Institute for Pediatric Surgical Innovation and Marshall Summar, M.D., director of the Children’s National Rare Disease Institute (CNRDI), can provide a more advanced diagnostic tool for regions of the world with limited access to geneticists or genetic testing.
The application utilizes artificial intelligence (AI) and machine learning to analyze biometric data and identify facial markers that are indicative of genetic disorders. Physicians can capture biometric data points of a child’s face in real time within the platform, where it scans facial biometric features to determine the potential presence of a genetic disease, which can often be life-threatening without early intervention. Research studies conducted in conjunction with the National Human Genome Research Institute at the National Institutes of Health further enhanced the development of the application in recent years, showing the potential to detect, with a 90 percent accuracy, early diagnosis of 128 genetic diseases across pediatric subjects in 28 countries. These diseases include DiGeorge syndrome (22q11.2 deletion syndrome), Down syndrome, Noonan syndrome and Williams-Beuren syndrome.
“We are delighted to enter into this licensing agreement through Innovation Ventures, the commercialization arm of Children’s National Hospital, which seeks to move inventions and discoveries from Children’s National to the marketplace to benefit the health and well-being of children. Our mission is to add the ‘D’ in development to the ‘R’ in research to accelerate the commercialization of our intellectual property,” says Kolaleh Eskandanian, Ph.D., M.B.A., P.M.P., vice president and chief innovation officer at Children’s National and managing director of Innovation Ventures. “It is through partnerships with startups and the industry that we can achieve this goal and thus we highly value this new partnership with MGeneRx Inc. The acceleration and commercialization of this objective digital biometric analysis technology will not only help diagnose rare genetic disorders – it will also allow for earlier interventions that improve the quality of life for the children living with these conditions.”
Eskandanian adds that the social impact of this technology is especially profound in lower income nations around the world, where there is a high prevalence of rare genetic conditions but a severe lack in the specialty care required to diagnose and treat them. Additional data collected through the expanded use of the technology will help to further develop the application and expand its capabilities to identify and diagnose additional rare genetic conditions.
The licensing agreement was arranged by the Children’s National Office of Innovation Ventures, which is focused on the commercialization of impactful new pediatric medical device technologies and therapies to advance children’s health care. Created to catalyze the ongoing translational research of the Children’s National Research Institute (CNRI) as well as inventions by hospital’s clinicians, Innovation Ventures focuses on four core pillars to advance pediatric medical technologies including a Biodesign program, partnerships and alliances to augment internal capacity, seed funding to de-risk technologies and validate market and clinical relevance, and back-office operations to manage intellectual property and licensing activities. Since 2017, Children’s National intellectual property has served as the basis for over 15 licensing or option agreements with commercial partners.
Providing access to an array of experts and resources for pediatric innovators is one of the aims of the Children’s National Research & Innovation Campus, a first-of-its-kind focused on pediatric health care innovation, with the first phase currently open on the former Walter Reed Army Medical Center campus in Washington, D.C. With its proximity to federal research institutions and agencies, universities, academic research centers, as well as on-site incubator Johnson and Johnson Innovation – JLABS, the campus provides a rich ecosystem of public and private partners, which will help bolster pediatric innovation and commercialization.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2017/12/Noonan-facial-recognition.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2021-07-14 15:48:322025-03-10 13:27:11Commercialization of novel facial analysis technology can improve diagnosis of rare disorders in pediatric patients
“The advent of different technologies and techniques over the years allowed pieces of her diagnosis to be made – and then brought all together,” says Andrew Dauber, M.D., MMSc.
After 20 years, a patient’s family received an answer to a decades-long genetic mystery. Their daughter had two rare disorders, Angelman syndrome and P450scc deficiency, which was detected after researchers found out she had uniparental disomy, two copies of chromosome 15 from one parent and none from another.
By using a variety of genetic tools, including whole-exome sequencing, microarray analyses and in-vitro modeling for gene splicing, the researchers were able to confirm this patient had uniparental disomy, a recessive genetic condition. They learned that after she received two impaired copies of chromosome 15 from her father, this woman developed a hormonal problem that led to adrenal insufficiency and sex reversal. This explained why she physically presented as a female, despite having testes and a Y-chromosome. It also explained other symptoms, including developmental delays and seizures.
“It’s a unique conglomeration of symptoms, manifested by the combination of these two very rare disorders,” says Andrew Dauber, M.D., MMSc., the division chief of endocrinology at Children’s National Health System and a guiding research author of this study. “The advent of different technologies and techniques over the years allowed pieces of her diagnosis to be made – and then brought together, commencing a 20-year diagnostic odyssey.”
For example, each of the conditions this patient has is known and rare: Angelman syndrome affects about one in 10 to 20,000 people in the U.S. Typical symptoms include those observed in this patient: delayed development, intellectual disability, speech impairment and seizures. Side-chain cleavage disorder, which leads to adrenal disorders and sex reversal, is also very rare. In 2005 the chances of survival with a P450scc defect were slim, but since then more than 28 infants have been diagnosed with this gene deficiency, which is required to convert cholesterol to pregnenolone, a hormone in the adrenal gland.
Dr. Dauber notes the chances of this occurring again are highly unlikely. The odds here are one in a gazillion. In this case, one disorder unmasked another, leaving researchers with new insights into the methodology for unraveling ultra-rare genetic disorders or for more common rare conditions.
“Knowing about the gene that caused the adrenal insufficiency and understanding this etiology won’t change medical care for this patient, but it will change the way researchers think about genetic detective work and about combining different technologies,” says Dr. Dauber. “We know that genetic disorders can be complex presentations of different disorders combined. This patient didn’t have one disorder, but three.”
When asked about the significance of the award, Dr. Dauber notes that, “It’s not that other people haven’t recognized this concept before, but this case is a striking example of it. Different technologies will unveil different types of genetic changes, which is why you have to use the right technology or the right technologies in the right combination to piece together the whole picture.”
Ahlee Kim, M.D., the lead study author and a clinical research fellow at Cincinnati Children’s Hospital Medical Center, will receive the award and the honorarium.
Additional study authors include Masanobu Fujimoto, Ph.D., Vivian Hwa, Ph.D., and Philippe Backeljauw, M.D., from Cincinnati Children’s Hospital.
The research was supported by grant K23HD07335, awarded to Dr. Dauber, from the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health (NIH). Additional funding included grant 1UL1TR001425 from the NIH’s National Center for Advancing Translational Sciences.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2019/04/DNA-Molecule.png300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2019-04-29 08:00:252020-10-12 09:19:07Using genomics to solve a 20-year case study
The growing popularity of genetic testing has one large hurdle: There are fewer than 4,000 genetic counselors in the United States, and people who use commercial genetic testing kits may receive confusing or inaccurate information.
To combat this problem, a team of doctors from the Rare Disease Institute at Children’s National Health System created the framework for a smartphone application that would house educational videos and tools that provide reputable information about genetic disorders and genetic testing.
On April 13, 2018, Debra Regier, M.D., Natasha Shur, M.D., and their teammates presented the app “Bear Genes” at the 2nd Annual Medical & Health App Development Workshop, a competition sponsored by the Clinical and Translational Science Institute at Children’s National (CTSI-CN) and the Milken Institute School of Public Health (Milken Institute SPH) at the George Washington University. Bear Genes won first place, and the team received $10,000 to develop a working prototype of the app.
The Bear Genes team was one of 10 who presented their ideas for smartphone apps to a panel of judges at the competition. Ideas covered a variety of topics, including emergency room visits and seizures related to menstrual cycles. Sean Cleary, Ph.D., M.P.H., an associate professor of epidemiology and biostatistics at the Milken Institute SPH, and his teammates proposed an app called “MyCommunicationPal” that would assist autistic individuals in reporting their symptoms to healthcare providers.
Sean Cleary and Kevin Cleary, Ph.D., technical director of the Bioengineering Initiative at Children’s National Health System, created the hackathon to bring together professionals from various fields to create technology-based solutions for public health and medical challenges. Interested participants submit applications and app proposals in the fall, and 10 ideas are selected to be fleshed out at the half-day hackathon. Participants join teams to develop the selected ideas, and on the day of the event, create a five-minute presentation to compete for the top prize. About 90 people attended this year’s hackathon.
“The workshop provides us with the opportunity to collaborate with healthcare providers, public health professionals and community members to develop an appropriate user-friendly app for those in need,” said Sean Cleary. “The event also fosters future collaborations between important stakeholders.”
This article originally appeared in the Milken Institute SPH pressroom.
https://innovationdistrict.childrensnational.org/wp-content/uploads/2018/04/2nd-annual-hackathon.jpg300400Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2018-04-23 14:37:272023-05-30 09:35:15Genetic testing reigns triumphant at health app hackathon
Using germline and tumor testing and centralized pathology review, a research team that included D. Ashley Hill, M.D, and Joyce Turner found that Sertoli-Leydig cell tumor and gynandroblastoma are nearly always DICER1-related tumors.
Children’s National Health System researchers played a key role in a new study exploring the clinical and genetic qualities of a group of rare, potentially deadly cancers that affect infants, children and adolescents. The research team’s findings suggest that genetic testing for people at risk may aid in earlier, more accurate diagnoses of these cancers, leading to early-stage treatment that could greatly improve survival.
Ovarian sex cord-stromal tumors (OSCST) include juvenile granulosa cell tumors (JGCT), Sertoli-Leydig cell tumor (SLCT) and gynandroblastoma (GAB). Mutations in the DICER1 gene often have been noted in children with these cancers, as well as in those with a particularly lethal pediatric lung cancer called pleuropulmonary blastoma (PPB). All of these cancers are highly curable if caught early but, at later stages, can be aggressive and often fatal.
Using germline and tumor testing and centralized pathology review, the research team found that SLCT and GAB are nearly always DICER1-related tumors. There also may be a much stronger association between SLCT and DICER1 than was previously appreciated. The new findings have implications for earlier detection and diagnoses of these cancers, as well as for screening other family members. The study was published in the December 2017 edition of Gynecologic Oncology.
“These types of tumors are diverse, relatively rare and understudied,” says D. Ashley Hill, M.D., the study’s senior author and a professor in the Division of Pathology and Laboratory Medicine at Children’s National. “Sertoli-Leydig cell tumor, for instance, is a unique genetic and pathologic entity and this rare cancer of the ovaries can be hard to detect. Using the testing process from this study, we now may be able to classify these tumors more accurately.”
The study authors assessed the first 107 individuals enrolled in the International Ovarian and Testicular Stromal Tumor Registry. They obtained medical and family history, and they conducted central pathology review plus DICER1 gene sequencing on blood and tumor tissue. Thirty-six of 37 patients with SLCTs and all four patients with GABs they tested showed DICER1 mutations, and half of those with SLCT had germline or mosaic mutations. The team noted that individuals with predisposing DICER1 mutations had significantly better overall and recurrence-free survival.
Based on their findings, the study authors recommend:
Careful and ideally centralized pathologic review for all individuals with OSCST tumors
DICER1 testing for all those with SLCT and GAB and
Consideration of DICER1 testing for patients with other OSCSTs.
“Genetic testing may be useful for screening and diagnosing entire families if one family member tests positive for a DICER1 mutation, especially to determine if they are at risk for PPB. When we know who is at risk, we can protect all children in a family,” Dr. Hill says. “Ultimately we may be able to cure this deadly lung cancer, PPB, by identifying and performing computed tomography scans on people who are at risk, so we can catch these cancers early.”
Dr. Hill thinks future research may study children whose cancer was not detected early or has become resistant to chemotherapy. They also may explore ways to restore normal controls in cancer cells, so they follow normal paths of development, for the purpose of developing targeted treatments with fewer side effects than current therapies.
In addition to Dr. Hill, other Children’s National study co-authors include Amanda Field, M.P.H., Department of Pathology; Weiying Yu, Ph.D., Department of Pathology; and Joyce Turner, director of the Cancer Genetic Counseling Program in Children’s Rare Disease Institute.
Other members of the study team are experts from the International Ovarian and Testicular Stromal Tumor Registry, Children’s Minnesota, Washington University Medical Center, Carolinas Health Care System, University of Texas MD Anderson Cancer Center, Harvard Medical School, University of Colorado School of Medicine, Clinic of Pediatrics (Dortmund, Germany), National Cancer Institute and Dana-Farber Cancer Institute.
Research reported in this story was supported by the National Institutes of Health under award number NCI R01CA143167, The Parson’s Foundation, St. Baldrick’s Foundation, Pine Tree Apple Tennis Classic Foundation, Hyundai Hope on Wheels, the Randy Shaver Cancer Research and Community Fund, the German Childhood Cancer Foundation and the Intramural Research Program of the Divisions of Cancer Epidemiology and Genetics, National Cancer Institute.
Changes or errors in an individual’s DNA are often at the root of many disorders. Personalized Sequencing is a fast, cost-effective way to look at a region of the genome without repeat tests and blood draws.
Until recently, doctors and patients had two choices for ordering genetic sequencing panels to identify underlying causes of disease—Individual Gene Testing (single genes and gene panels) or Whole Exome Sequencing.
Individual gene testing is the standard testing modality. Physicians identify a single gene to analyze for change or mutation. If results are negative, they order another individual test, requiring a repeat visit and another blood draw. The process is repeated again and again based on likely candidate genes for a specific disease or symptom. If a physician is very lucky, it takes only a few rounds of tests to find the culprit. More likely, however, the number of individual tests grows large, taking months of patients’ time and increasing healthcare costs significantly. By contrast, Whole Exome Sequencing includes sequencing and analyses of 25,000 genes. It is more expensive when compared with individual gene testing and takes three to six months to complete. When complete, the results often can be more than the doctor and patient bargained for: Potentially revealing a genetic problem that is unrelated to the patient’s current symptoms. A 3-year-old with seizures also may come up positive for BRCA1, the breast cancer gene. Knowing that doesn’t help understand what causes the seizures or how to best treat them. In this model, you receive everything you could ever want. Because there is so much information, however, the results are difficult to interpret or to inform treatment decisions.
We’ve come up with a different way: Personalized Sequencing Panels, a precision medicine initiative at Children’s National Health System. We offer physicians a menu of genetic regions from which to choose when they order a sequencing analysis. While a medical exome is still sequenced, we only analyze a subset of genes that the physician and geneticist think are the most likely targets, which reduces the cost and time for analysis compared to Whole Exome Sequencing. Targeting regions in this approach shortens our turnaround time for results to two or three weeks. If the first identified region shows nothing, we can return to data we’ve already collected for a second look.
We’ve been using the model for 18 months and have tested more than 1,000 patients this way. Eighty percent of physicians prefer to “create their own test” using our menu of options. Rather than bringing a one-size-fits-all test to the patient, we bring the patient their very own personalized test.
About the Author
Sean Hofherr Laboratory Medicine Specialist
https://innovationdistrict.childrensnational.org/wp-content/uploads/2016/09/PerspectivesImage_Personalized-Sequencing-Tailors_sm-2.jpg402600Innovation Districthttps://innovationdistrict.childrensnational.org/wp-content/uploads/2023/12/innovationdistrict_logo-1-1030x165.pngInnovation District2016-08-06 18:12:362017-08-15 10:05:25Personalized sequencing tailors genetic tests for each patient








 Sean Hofherr
Sean Hofherr