Study suggests glioblastoma tumors originate far from resulting tumors

“The more we continue to learn about glioblastoma,” Yuan Zhu, Ph.D., says, “the more hope we can give to these patients who currently have few effective options.”
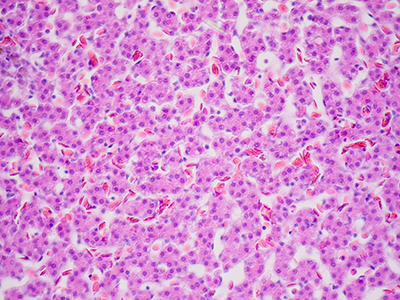
A pre-clinical model of glioblastoma, an aggressive type of cancer that can occur in the brain, suggests that this recalcitrant cancer originates from a pool of stem cells that can be a significant distance away from the resulting tumors. The findings of a new study, led by Children’s National Hospital researchers and published July 22 in the journal Nature Communications, suggest new ways to fight this deadly disease.
Despite decades of research, glioblastoma remains the most common and lethal primary brain tumor in adults, with a median survival of only 15 months from diagnosis, says study leader Yuan Zhu, Ph.D., the scientific director and endowed professor of the Gilbert Family Neurofibromatosis Institute at Children’s National. Unlike many cancers, which start out as low-grade tumors that are more treatable when they’re caught at an early stage, most glioblastomas are almost universally discovered as high-grade and aggressive lesions that are difficult to treat with the currently available modalities, including surgery, radiation and chemotherapy.
“Once the patient has neurological symptoms like headache, nausea, and vomiting, the tumor is already at an end state, and disease progression is very rapid,” Dr. Zhu says. “We know that the earlier you catch and treat cancers, the better the prognosis will be. But here, there’s no way to catch the disease early.”
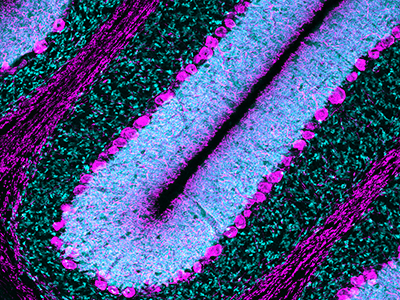
However, some recent research in glioblastoma patients shows that the subventricular zone (SVZ) – an area that serves as the largest source of stem cells in the adult brain – contains cells with cancer-driving mutations that are shared with tumors found in other often far-distant brain regions.
To see if the SVZ might be the source for glioblastoma tumors, Dr. Zhu and his colleagues worked with pre-clinical models that carried a single genetic glitch: a mutation in a gene known as p53 that typically suppresses tumors. Mutations in p53 are known to be involved in glioblastoma and many other forms of cancer.
Using genetic tests and an approach akin to those used to study evolution, the researchers traced the cells that spurred both kinds of tumors back to the SVZ. Although both single and multiple tumors had spontaneously acquired mutations in a gene called Pten, another type of tumor suppressor, precursor cells for the single tumors appeared to acquire this mutation before they left the SVZ, while precursor cells for the multiple tumors developed this mutation after they left the stem cell niche. When the researchers genetically altered the animals to shut down the molecular pathway that loss of Pten activates, it didn’t stop cancer cells from forming. However, rather than migrate to distal areas of the brain, these malignant cells remained in the SVZ.
Dr. Zhu notes that these findings could help explain why glioblastoma is so difficult to identify the early precursor lesions and treat. This work may offer potential new options for attacking this cancer. If new glioblastoma tumors are seeded by cells from a repository in the SVZ, he explains, attacking those tumors won’t be enough to eradicate the cancer. Instead, new treatments might focus on this stem cell niche as target for treatment or even a zone for surveillance to prevent glioblastoma from developing in the first place.
Another option might be to silence the Pten-suppressed pathway through drugs, a strategy that’s currently being explored in various clinical trials. Although these agents haven’t shown yet that they can stop or reverse glioblastomas, they might be used to contain cancers in the SVZ as this strategy did in the pre-clinical model — a single location that might be easier to attack than tumors in multiple locations.
“The more we continue to learn about glioblastoma,” Dr. Zhu says, “the more hope we can give to these patients who currently have few effective options.”
Other Children’s National researchers who contributed to this study include Yinghua Li, Ph.D., Wei Li, Ph.D., Yuan Wang, Ph.D., Seckin Akgul, Ph.D., Daniel M. Treisman, Ph.D., Brianna R. Pierce, B.S., Cheng-Ying Ho, M.D. /Ph.D.
This work is supported by grants from the National Institutes of Health (2P01 CA085878-10A1, 1R01 NS053900 and R35CA197701).