Molecular gatekeepers that regulate calcium ions key to muscle function
Controlled entry of calcium ions into the mitochondria, the cell’s energy powerhouses, makes the difference between whether muscles grow strong or easily tire and perish from injury, according to research published in Cell Reports.
Calcium ions are essential to how muscles work effectively, playing a starring role in how and when muscles contract, tap energy stores to keep working and self-repair damage. Not only are calcium ions vital for the repair of injured muscle fibers, their controlled entry into the mitochondria, the cell’s energy powerhouses, spells the difference between whether muscles will be healthy or if they will easily tire and perish following an injury, according to research published Oct. 29, 2019, in Cell Reports.
“Lack of the protein mitochondrial calcium uptake1 (MICU1) lowers the activation threshold for calcium uptake mediated by the mitochondrial calcium uniporter in both, muscle fibers from an experimental model and fibroblast of a patient lacking MICU1,” says Jyoti K. Jaiswal, MSc, Ph.D., a principal investigator in the Center for Genetic Medicine Research at Children’s National Hospital and one of the paper’s corresponding authors. “Missing MICU1 also tips the calcium ion balance in the mitochondria when muscles contract or are injured, leading to more pronounced muscle weakness and myofiber death.”
Five years ago, patients with a very rare disease linked to mutations in the mitochondrial gene MICU1 were described to suffer from a neuromuscular disease with signs of muscle weakness and damage that could not be fully explained.
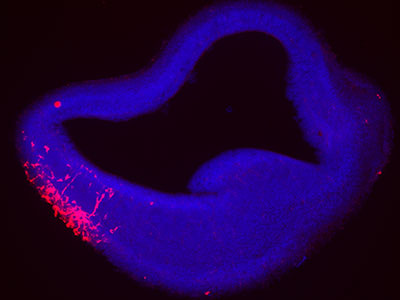
To determine what was going awry, the multi-institutional research team used a comprehensive approach that included fibroblasts donated by a patient lacking MICU1 and an experimental model whose MICU1 gene was deleted in the muscles.
Loss of MICU1 in skeletal muscle fibers leads to less contractile force, increased fatigue and diminished capacity to repair damage to their cell membrane, called the sarcolemma. Just like human patients, the experimental model suffers more pronounced muscle weakness, increased numbers of dead myofibers, with greater loss of muscle mass in certain muscles, like the quadriceps and triceps, the research team writes.
“What was happening to the patient’s muscles was a big riddle that our research addressed,” Jaiswal adds. “Lacking this protein is not supposed to make the muscle fiber die, like we see in patients with this rare disease. The missing protein is just supposed to cause atrophy and weakness.”
Patients with this rare disease show early muscle weakness, fluctuating levels of fatigue and lethargy, muscle aches after exercise, and elevated creatine kinase in their bloodstream, an indication of cell damage due to physical stress.
“One by one, we investigated these specific features in experimental models that look normal and have normal body weight, but also show lost muscle mass in the quadriceps and triceps,” explains Adam Horn, Ph.D., the lead researcher in Jaiswal’s lab who conducted this study. “Our experimental model lacking MICU1 only in skeletal muscles responded to muscle deficits so similar to humans that it suggests that some of the symptoms we see in patients can be attributed to MICU1 loss in skeletal muscles.”
Future research will aim to explore the details of how the impact of MICU1 deficit in muscles may be addressed therapeutically and possible implications of lacking MICU1 or its paralog in other organs.
In addition to Jaiswal and Horn, Children’s National Hospital Center for Genetic Medicine Research co-authors include Marshall W. Hogarth and Davi A. Mazala. Additional co-authors include Lead Author Valentina Debattisti, Raghavendra Singh, Erin L. Seifert, Kai Ting Huang, and Senior Author György Hajnóczky, all from Thomas Jefferson University; and Rita Horvath, from Newcastle University.
Financial support for research described in this post was provided by the National Institutes of Health under award numbers R01AR55686, U54HD090257 and RO1 GM102724; National Institute of Arthritis and Musculoskeletal and Skin Diseases under award number T32AR056993; and Foundation Leducq.