Special issue of “Neurochemical Research” honors Vittorio Gallo, Ph.D.

Investigators from around the world penned manuscripts that were assembled in a special issue of “Neurochemical Research” that honors Vittorio Gallo, Ph.D., for his leadership in the field of neural development and regeneration.
At a pivotal moment early in his career, Vittorio Gallo, Ph.D., was accepted to work with Professor Giulio Levi at the Institute for Cell Biology in Rome, a position that leveraged courses Gallo had taken in neurobiology and neurochemistry, and allowed him to work in the top research institute in Italy directed by the Nobel laureate, Professor Rita Levi-Montalcini.
For four years as a student and later as Levi’s collaborator, Gallo focused on amino acid neurotransmitters in the brain and mechanisms of glutamate and GABA release from nerve terminals. Those early years cemented a research focus on glutamate neurotransmission that would lead to a number of pivotal publications and research collaborations that have spanned decades.
Now, investigators from around the world who have worked most closely with Gallo penned tributes in the form of manuscripts that were assembled in a special issue of “Neurochemical Research” that honors Gallo “for his contributions to our understanding of glutamatergic and GABAergic transmission during brain development and to his leadership in the field of neural development and regeneration,” writes guest editor Arne Schousboe, of the University of Copenhagen in Denmark.

Vittorio Gallo, Ph.D. as a 21-year-old mustachioed graduate student.
“In spite of news headlines about competition in research and many of the negative things we hear about the research world, this shows that research is also able to create a community around us,” says Gallo, chief research officer at Children’s National Hospital and scientific director for the Children’s National Research Institute.
As just one example, he first met Schousboe 44 years ago when Gallo was a 21-year-old mustachioed graduate student.
“Research can really create a sense of community that we carry on from the time we are in training, nurture as we meet our colleagues at periodic conferences, and continue up to the present. Creating community is bi-directional: influencing people and being influenced by people. People were willing to contribute these 17 articles because they value me,” Gallo says. “This is a lot of work for the editor and the people who prepared papers for this special issue.”
In addition to Gallo publishing more than 140 peer-reviewed papers, 30 review articles and book chapters, Schousboe notes a number of Gallo’s accomplishments, including:
- He helped to develop the cerebellar granule cell cultures as a model system to study how electrical activity and voltage-dependent calcium channels modulate granule neuron development and glutamate release.
- He developed a biochemical/neuropharmacological assay to monitor the effects of GABA receptor modulators on the activity of GABA chloride channels in living neurons.
- He and Maria Usowicz used patch-clamp recording and single channel analysis to demonstrate for the first time that astrocytes express glutamate-activated channels that display functional properties similar to neuronal counterparts.
- He characterized one of the spliced isoforms of the AMPA receptor subunit gene Gria4 and demonstrated that this isoform was highly expressed in the cerebellum.
- He and his Children’s National colleagues demonstrated that glutamate and GABA regulate oligodendrocyte progenitor cell proliferation and differentiation.
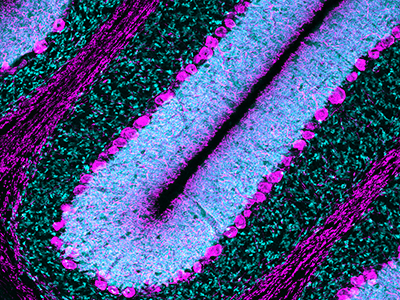
Purkinje cells are large neurons located in the cerebellum that are elaborately branched like interlocking tree limbs and represent the only source of output for the entire cerebellar cortex.
Even the image selected to grace the special issue’s cover continues the theme of continuity and leaving behind a legacy. That image of Purkinje cells was created by a young scientist who works in Gallo’s lab, Aaron Sathyanesan, Ph.D. Gallo began his career working on the cerebellum – a region of the brain important for motor control – and now studies with a team of scientists and clinician-scientists Purkinje cells’ role in locomotor adaptive behavior and how that is disrupted after neonatal brain injury.
“These cells are the main players in cerebellar circuitry,” Gallo says. “It’s a meaningful image because goes back to my roots as a graduate student and is also an image that someone produced in my lab early in his career. It’s very meaningful to me that Aaron agreed to provide this image for the cover of the special issue.”