Experimental model mimics early-stage myogenic deficit in boys with DMD
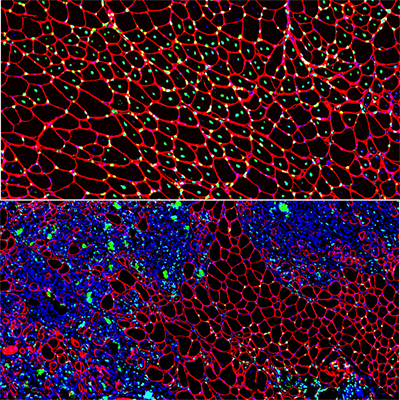
Muscle regeneration marked by incorporation of muscle stem cell nuclei (green) in the myofibers (red) in dystrophic muscles with low TGFβ level (upper image), but not with high TGFβ level (lower image). Inflammatory and other nuclei are labeled blue.
Boys with Duchenne muscular dystrophy (DMD) experience poor muscle regeneration, but the precise reasons for this remain under investigation. An experimental model of severe DMD that experiences a large spike in transforming growth factor-beta (TGFβ) activity after muscle injury shows that high TGFβ activity suppresses muscle regeneration and promotes fibroadipogenic progenitors (FAPs). This leads to replacement of the damaged muscle fibers by calcified and connective tissue, compromising muscle structure and function. While blocking FAP buildup provides a partial solution, a Children’s National Hospital study team identifies correcting the muscle micro-environment caused by high TGFβ as a ripe therapeutic target.
The team’s study was published online March 26, 2020, in JCI Insight.
DMD is a chronic muscle disease that affects 1 in 6,200 young men in the prime of their lives. The disorder, caused by genetic mutations leading to the inability to produce dystrophin protein, leads to ongoing muscle damage, chronic inflammation and poor regeneration of lost muscle tissue. The patients experience progressive muscle wasting, lose the ability to walk by the time they’re teenagers and die prematurely due to cardiorespiratory failure.
The Children’s National team finds for the first time that as early as preadolescence (3 to 4 weeks of age), their experimental model of severe DMD disease showed clear signs of the type of spontaneous muscle damage, regenerative failure and muscle fiber loss seen in preadolescent boys who have DMD.
“In boys, the challenge due to muscle loss exists from early in their lives, but had not been mimicked previously in experimental models,” says Jyoti K. Jaiswal, MSc, Ph.D., principal investigator in the Center for Genetic Medicine Research at Children’s National, and the study’s co-senior author. “TGFβ is widely associated with muscle fibrosis in DMD, when, in fact, our work shows its role in this disease process is far more significant.”
Research teams have searched for experimental models that replicate the sudden onset of symptoms in boys who have DMD as well as its complex progression.
“Our work not only offers insight into the delicate balance needed for regeneration of skeletal muscle, but it also provides quantitative information about muscle stem cell activity when this balanced is disturbed,” says Terence A. Partridge, Ph.D., principal investigator in the Center for Genetic Medicine Research at Children’s National, and the study’s co-senior author.
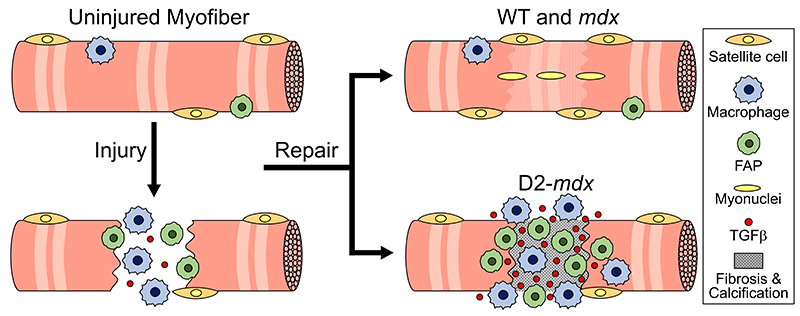
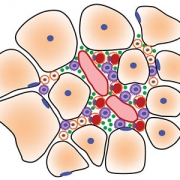
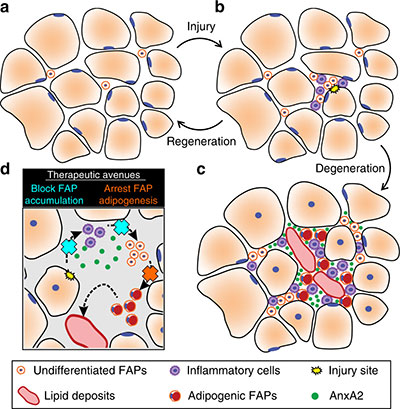
This schematic depicts the fate of injured myofibers in healthy or dystrophic muscle (WT or mdx experimental models) that maintain low TGFβ level, compared with D2-mdx experimental models that experience a large increase in TGFβ level. As the legend shows, various cells are involved in this regenerative response.
“The D2-mdx experimental model is a relevant one to use to investigate the interplay between inflammation and muscle degeneration that is seen in humans with DMD,” adds Davi A.G. Mázala, co-lead study author. “This model faithfully recapitulates many features of the complex disease process seen in humans.”
Between 3 to 4 weeks of age in the experimental models of severe DMD disease, the level of active TGFβ spiked up to 10-fold compared with models with milder disease. Intramuscular injections of an off-the-shelf drug that inhibits TGFβ signaling tamped down the number of FAPs, improving the muscle environment by lowering TGFβ activity.
“This work lays the foundation for studies that could lead to future therapeutic strategies to improve patients’ outcomes and lessen disease severity,” says James S. Novak, Ph.D., principal investigator in Children’s Center for Genetic Medicine Research, and co-lead study author. “Ultimately, our goal is to improve the ability of patients to continue to maintain muscle mass and regenerate muscle.”
In addition to Mázala, Novak, Jaiswal and Partridge, Children’s National study co-authors include Marshall W. Hogarth; Marie Nearing; Prabhat Adusumalli; Christopher B. Tully; Nayab F. Habib; Heather Gordish-Dressman, M.D.; and Yi-Wen Chen, Ph.D.
Financial support for the research described in this post was provided by the National Institutes of Health under award Nos. T32AR056993, R01AR055686 and U54HD090257; Foundation to Eradicate Duchenne; Muscular Dystrophy Association under award Nos. MDA295203, MDA480160 and MDA 477331; Parent Project Muscular Dystrophy; and Duchenne Parent Project – Netherlands.