ER maintains ion balance needed for muscle repair
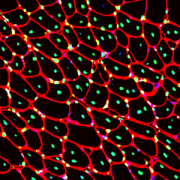
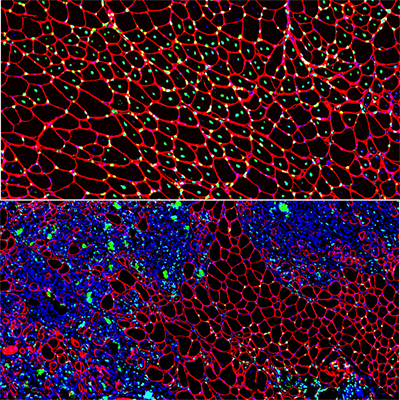
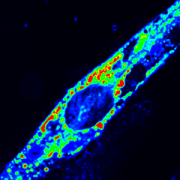
A new study led by Jyoti Jaiswal, M.Sc., Ph.D., principal investigator at Children’s National Hospital, identifies that an essential requirement for the repair of injured cells is to cope with the extracellular calcium influx caused by injury to the cell’s membrane. Credit: Goutam Chandra, Ph.D.
Physical activity can injure our muscle cells, so their ability to efficiently repair is crucial for maintaining muscle health. Understanding how healthy muscle cells respond to injury is required to understand and treat diseases caused by poor muscle cell repair.
A new study led by Jyoti Jaiswal, M.Sc., Ph.D., principal investigator at Children’s National Hospital, identifies that an essential requirement for the repair of injured cells is to cope with the extracellular calcium influx caused by injury to the cell’s membrane.
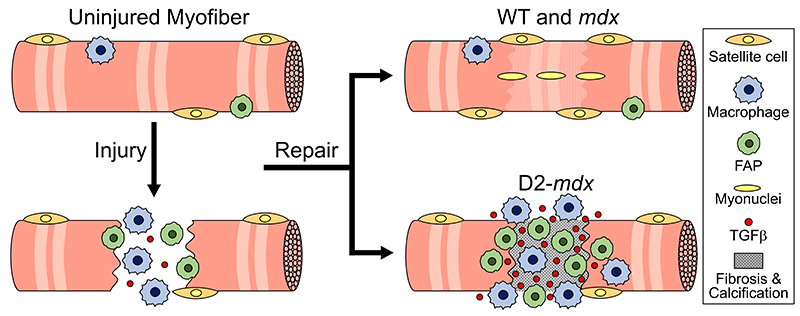
This study, published in the Journal of Cell Biology, identifies endoplasmic reticulum (ER) – a network of membranous tubules in the cell – as the site where the calcium entering the injured cell is sequestered. Using limb girdle muscular dystrophy 2L (LGMD2L) patient cells and a model for this genetic disease, the study shows impaired ability of diseased muscle cells to cope with this calcium excess. It also shows that a drug to sequester excess calcium counters this ion imbalance and reverses the diseased cell’s repair deficit.
“The study provides a novel insight into how injured cells in our body cope with calcium ion imbalance during injury,” Dr. Jaiswal explained. “This work also addresses how calcium homeostasis is compromised by a genetic defect that leads to LGMD2L. It also offers a proof of principle approach to restore calcium homeostasis, paving the path for future work to develop therapies targeting this disease.”
According to Dr. Jaiswal, this work also addresses the current lack of understanding of the basis for exercise intolerance and other symptoms faced by LGMD2L patients.
“This study opens the path for developing targeted therapies for LGMD2L and provides a fundamental cellular insight into a process crucial for cell survival,” said Goutam Chandra, Ph.D., research fellow and lead author of this study.
The Center for Genetic Medicine Research at Children’s National is among only a handful across the world to study this rare disease. These findings are unprecedented in providing the mechanistic insights needed to develop treatment for it.
In addition to Dr. Jaiswal and Chandra, the study co-authors include Sreetama Sen Chandra, Ph.D., Davi Mazala, Ph.D., and Jack VanderMeulen, Ph.D., from Children’s National, and Karine Charton, Ph.D., and Isabelle Richard, Ph.D., from Université Paris-Saclay.