Reducing urinary protein for patients with FSGS slows kidney decline

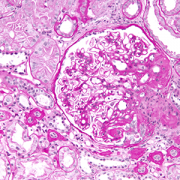
High magnification micrograph of focal segmental glomerulosclerosis (FSGS).
Reducing the amount of protein in the urine of patients with focal segmental glomerulosclerosis (FSGS), a rare disease in which scar tissue forms on the parts of the kidneys that filter waste from the blood, can significantly slow declines in kidney function and extend time before patients’ kidneys fail, a new analysis by a Children’s National Hospital researcher and her colleagues shows. These findings, published online Aug. 10, 2020, in the American Journal of Kidney Disease, could provide hope for patients who are able to lower their urinary protein with available treatments but aren’t able to achieve complete remission, the researchers say.
FSGS affects about seven per every million people in the general population. However, in the United States, it’s responsible for between 5 and 20% of all cases of end stage kidney disease (ESKD), a condition in which the kidney function declines enough that patients can’t survive without dialysis or a kidney transplant. There are no proven treatments specifically targeting FSGS, but steroids and other immunosuppressants have shown promise in clinical trials.
One characteristic sign of FSGS is proteinuria, in which too much protein is present in patients’ urine. Most clinical trials of FSGS treatments have focused on complete remission of proteinuria as a sign that the intervention is working. However, says Marva Moxey-Mims, M.D., researcher and chief of the Children’s National Division of Nephrology, only a fraction of patients meet that end goal. Instead, many patients achieve some reduction in proteinuria, but it’s been unclear whether those reductions lead to significant benefits for kidney health.
To investigate this question, Dr. Moxey-Mims and her colleagues used data from the National Institutes of Health-funded FSGS clinical trial that took place between November 2004 and May 2008. Participants in this study — 138 patients who developed proteinuria due to FSGS between the ages of 2 and 40 and didn’t respond to steroids — received one of two different immunosuppressant regimens. They received frequent checkups including urinary protein tests during the duration of the study and were followed for a maximum of 54 months.
Results showed that about 49% of the study participants’ proteinuria improved by 26 weeks of treatment on either regimen. More importantly, says Dr. Moxey-Mims, these patients retained significantly better kidney function over time, determined by a test called estimated glomerular filtration rate (eGFR), compared to those whose urinary protein remained high. The greater the reduction in proteinuria, the better their kidney function remained, and the longer their kidneys remained active before they developed ESKD.
“Even a modest reduction in proteinuria, as small as 20 or 30%, had an impact on these patients’ kidney health,” Dr. Moxey-Mims says.
Dr. Moxey-Mims notes that the finding could impact the design of clinical trials for FSGS treatments. Currently, these trials typically must include large numbers of patients to show a benefit if complete remission of proteinuria — which only occurred in about 20% of patients in the National Institute of Diabetes and Digestive and Kidney Diseases trial — is used as the end point.
If researchers use a range of proteinuria reduction as end points, she says, it could be easier to see if a drug or other intervention is working.
Similarly, she says, patients with FSGS and their doctors should view any proteinuria reduction as a positive.
“They shouldn’t be discouraged if they can’t reach full remission,” Dr. Moxey-Mims says. “Doctors and patients alike can feel reassured that if they’re reducing protein in the urine to some degree, then patients are getting some benefit.”