Gene therapy offers potential long-term treatment for limb-girdle muscular dystrophy 2B
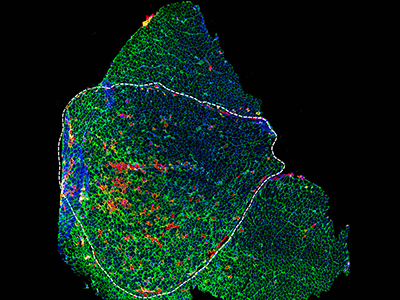
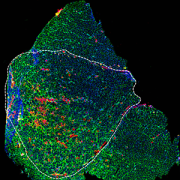
Microscopic visual of a diseased muscle section. Credit: Daniel Bittel.
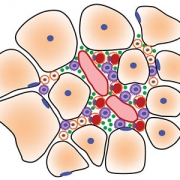
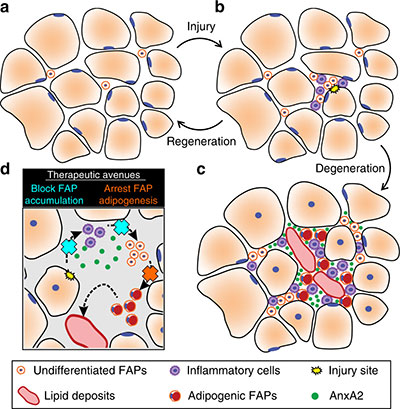
Children’s National Hospital experts developed a new pre-clinical gene therapy for a rare disorder, known as limb-girdle muscular dystrophy (LGMD) 2B, that addresses the primary cellular deficit associated with this disease. Using a single injection of a low dose gene therapy vector, researchers restored the ability of injured muscle fibers to repair in a way that reduced muscle degeneration and enhanced the functioning of the diseased muscle. The treatment was safe, attenuated fibro-fatty muscle degeneration, and restored myofiber size and muscle strength, according to the study published in the Journal of Clinical Investigation.
With an incidence of less than 1 in 100,000, LGMD2B is a rare disorder caused by a genetic mutation in a large gene called dysferlin. This faulty gene leads to muscle weakness in the arms, legs, shoulder and pelvic girdle. Affected children and adults face trouble walking, climbing stairs and getting out of chairs. Individuals typically lose the ability to walk within years after the onset of symptoms, and often need assistance with everyday tasks such as showering, dressing and transferring.
This study described a new approach that avoids the need for packaging a large gene, like dysferlin, or giving a large vector dose to target the muscles, which are bottlenecks faced in ongoing gene therapy efforts aimed at muscular dystrophies.
“Currently, patients with LGMD2B have no gene or drug-based therapies available to them, and we are amongst the few centers developing therapeutic approaches for this disease,” said Jyoti K. Jaiswal, M.Sc. Ph.D., senior investigator of the Center for Genetic Medicine Research at Children’s National. “We are working to further enhance the efficacy of this approach and perform a longer-term safety and efficacy study to enable the clinical translation of this therapy.”
The genetic defect in dysferlin that is associated with LGMD2B causes the encoded protein to be truncated or degraded. This hinders the muscle fiber’s ability to heal, which is required for healthy muscles. In recessive genetic disorders, like LGMD2B, common pre-clinical gene therapy approaches usually target the mutated gene in the muscle, making them capable of producing the missing proteins.
“The large size of the gene mutated in this disease, and impediments in body-wide delivery of gene therapy vectors to reach all the muscles, pose significant challenges for developing gene therapies to treat this disease,” said Jaiswal.
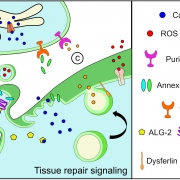
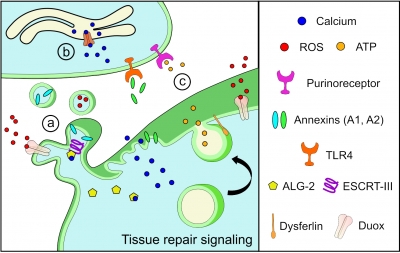
To overcome these challenges, the researchers found another way to slow down the disease’s progression. The authors built upon their previous discovery that acid sphingomyelinase (hASM) protein is required to repair injured muscle cells. In this current work, the research team administered a single in vivo dose of an Adeno-associated virus (AAV) vector that produces a secreted version of hASM in the liver, which then was delivered to the muscles via blood circulation at a level determined to be efficacious in repairing LGMD2B patient’s injured muscle cells.
“Increased muscle degeneration necessitates greater muscle regeneration, and we found that improved repair of dysferlin-deficient myofibers by hASM-AAV reduces the need for regeneration, causing a 2-fold decrease in the number of regenerated myofibers,” said Daniel Bittel, D.P.T., PhD., research postdoctoral fellow of the Center for Genetic Medicine Research at Children’s National and a lead author of this study.
Sreetama Sen Chandra, Ph.D., who was a research postdoctoral fellow at Children’s National at the time of this study and served as co-lead author, also added that “these findings are also of interest to patients with Niemann-Pick disease type A since the pre-clinical model for this disease also manifests poor sarcolemma repair.”
Children’s National researchers of the Center for Genetic Medicine Research and the Rare Disease Institute (RDI) are constantly pursuing high-impact opportunities in pediatric genomic and precision medicine. Both centers combine its strengths with public and private partners, including industry, universities, federal agencies, start-up companies and academic medical centers. They also serve as an international referral site for rare disorders.
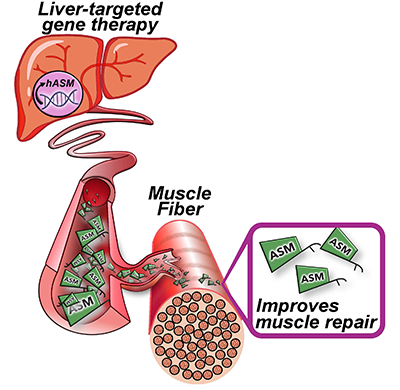
Gene therapy Schematic. Credit: Daniel Bittel.