Losing muscle to fat: misdirected fate of a multipotent stem cell drives LGMD2B
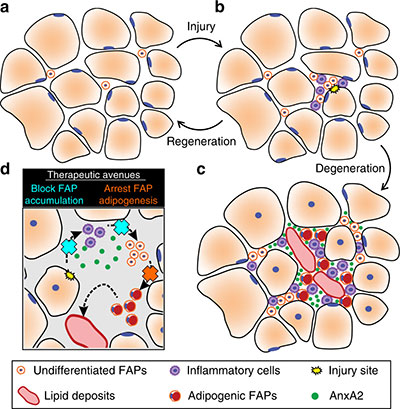
Fibro/adipogenic precursors (FAPs) control the onset and severity of disease in limb-girdle muscular dystrophy type 2 (LGMD2B). a) Healthy and/or pre-symptomatic LGMD2B muscle contains resident FAPs. b) After myofiber injury, inflammatory cells invade and trigger FAP proliferation. c) In symptomatic LGMD2B muscle, there is a gradual accumulation of extracellular AnxA2, which prolongs the pro-inflammatory environment, causing excessive FAP proliferation. d) Blocking aberrant signaling due to AnxA2 buildup blocks FAP accumulation and thus preventing adipogenic loss of dysferlinopathic muscle. Credit: “Fibroadipogenic progenitors are responsible for muscle loss in limb girdle muscular dystrophy 2B.” Published online June 3, 2019, in Nature Communications. Marshall W. Hogarth, Aurelia Defour, Christopher Lazarski, Eduard Gallardo, Jordi Diaz Manera, Terence A. Partridge, Kanneboyina Nagaraju and Jyoti K. Jaiswal. https://rdcu.be/bFu9U.
Research led by faculty at Children’s National published online June 3, 2019, in Nature Communications shows that the sudden appearance of symptoms in limb-girdle muscular dystrophy type 2 (LGMD2B) is a result of impaired communication between different cell types that facilitate repair in healthy muscle. Of particular interest are the fibro/adipogenic precursors (FAPs), cells that typically play a helpful role in regenerating muscle after injury by removing debris and enhancing the fusion of muscle cells into new myofibers.
LGMD2B is caused by mutations in the DYSF gene that impair the function of dysferlin, a protein essential for repairing injured muscle fibers. Symptoms, like difficulty climbing or running, do not appear in patients until young adulthood. This late onset has long puzzled researchers, as the cellular consequences of dysferlin’s absence are present from birth and continue through development, but do not impact patients until later in life.
The study found that in the absence of dysferlin, muscle gradually increases the expression of the protein Annexin A2 which, like dysferlin, facilitates repair of injured muscle fiber. However, increasing Annexin A2 accumulates outside the muscle fiber and drives an increase in FAPs within the muscle as well as encourages these FAPs to differentiate into adipocytes, forming fatty deposits. Shutting down Annexin A2 or blocking the adipocyte fate of FAPs using an off-the-shelf medicine arrests the fatty replacement of dysferlinopathic muscle.
“We propose a feed-forward loop in which repeated myofiber injury triggers chronic inflammation which, over time, creates an environment that promotes FAPs to accumulate and differentiate into fat. This, in turn, contributes to more myofiber damage,” says Jyoti K. Jaiswal, MSc, Ph.D., a principal investigator in the Center for Genetic Medicine Research at Children’s National and the study’s senior author.
“Adipogenic accumulation becomes the nucleating event that results in an abrupt decline in muscle function in patients. This new view of LGMD2B disease opens previously unrealized avenues to intervene,” adds Marshall Hogarth, Ph.D., the study’s lead author.

“We propose a feed-forward loop in which repeated myofiber injury triggers chronic inflammation which, over time, creates an environment that promotes FAPs to accumulate and differentiate into fat. This, in turn, contributes to more myofiber damage,” says Jyoti K. Jaiswal, MSc, Ph.D.
A research team led by Jaiswal collaborated with Eduard Gallardo and Jordi Diaz Manera, of Hospital de la Santa Creu in Barcelona, Spain, to examine muscle biopsies from people with LGMD2B who had mild to severe symptoms. They found that adipogenic deposits originate in the extracellular matrix space between muscle fibers, with the degree of accumulation tied to disease severity. They found a similar progressive increase in lipid accumulation between myofibers predicted disease severity in dysferlin-deficient experimental models. What’s more, this process can be accelerated by muscle injury, triggering increased adipogenic replacement in areas that otherwise would be occupied by muscle cells.
“Accumulation and adipogenic differentiation of FAPs is responsible for the decline in function for dysferlinopathic muscle. Reversing this could provide a therapy for LGMD2B, a devastating disease with no effective treatment,” predicts Jaiswal as the team continues research in this field.
Promising off-the-shelf drugs include batimastat, an anti-cancer drug that inhibits the extracellular matrix enzyme matrix metalloproteinase. This drug reduces FAP adipogenesis in vitro and lessens injury-triggered lipid formation in vivo. In experimental models, batimastat also increases muscle function.
In addition to Jaiswal, Hogarth, Gallardo and Diaz Manera, other study co-authors include Aurelia Defour, Christopher Lazarski, Terence A. Partridge and Kanneboyina Nagaraju, all of Children’s National.
Financial support for research described in this post was provided by the Muscular Dystrophy Association under awards MDA477331 and MDA277389, the National Institute of Arthritis and Musculoskeletal and Skin Diseases under award R01AR055686 and the National Institutes of Health under awards K26OD011171, R24HD050846 and P50AR060836.



